
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Mitochondrial Dysfunction as a Pathophysiological Bridge between Metabolic Dysfunction-Associated Fatty Liver Disease and Chronic Kidney Disease
1 Department of Pediatrics, The Affiliated Wuxi People’s Hospital of Nanjing Medical University, Wuxi People’s Hospital, Wuxi Medical Center, Nanjing Medical University, Wuxi, 214023, China
2 Department of Cardiology, The Affiliated Wuxi People’s Hospital of Nanjing Medical University, Wuxi People’s Hospital, Wuxi Medical Center, Nanjing Medical University, Wuxi, 214023, China
3 Department of Nephrology, Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 200127, China
4 Department of Pediatric Laboratory, Affiliated Children’s Hospital of Jiangnan University, Wuxi Children’s Hospital, Wuxi, 214023, China
* Corresponding Authors: Le Zhang. Email: ; Xiaolei Wang. Email:
; Yutong Hou. Email:
# These authors contributed equally to this work as the first author
BIOCELL 2026, 50(3), 2 https://doi.org/10.32604/biocell.2025.072971
Received 08 September 2025; Accepted 14 November 2025; Issue published 23 March 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Metabolic dysfunction-associated fatty liver disease (MAFLD) and chronic kidney disease (CKD) have shown a marked global increase in prevalence, placing a substantial burden on public health and healthcare systems worldwide. Epidemiological data demonstrate a significant overlap between these two conditions, with further evidence from research identifying common pathophysiological features, such as lipid metabolism dysregulation, disrupted energy balance, and chronic systemic inflammation. Mitochondria are central to the pathophysiology of both diseases. In addition to their role in energy production, mitochondria are involved in numerous critical cellular processes, including biosynthesis, lipid metabolism, oxidative phosphorylation, signal transduction, and apoptosis regulation. Mitochondrial dysfunction, characterized by increased reactive oxygen species, impaired adenosine triphosphate synthesis, disrupted mitophagy, and changes in mitochondrial morphology, is implicated in the progression of both MAFLD and CKD. Given the pivotal role of mitochondria in maintaining cellular metabolism homeostasis, dysfunction of this organelle is increasingly recognized as a key mechanistic link that connects the pathophysiological processes underlying both MAFLD and CKD. This review underscores mitochondrial dysfunction as a pathogenic nexus between MAFLD and CKD and examines the mechanisms that drive their pathogenesisKeywords
Metabolic dysfunction-associated fatty liver disease (MAFLD) and chronic kidney disease (CKD) are increasingly recognized as major metabolic disorders, with expanding prevalence that poses significant challenges to public health worldwide [1,2]. Non-alcoholic fatty liver disease (NAFLD) refers to the excessive accumulation of fat in hepatocytes, which can be diagnosed only after excluding other clear causes of liver damage, such as excessive alcohol consumption [3]. Patients with NAFLD may progress from simple hepatic steatosis to non-alcoholic steatohepatitis (NASH), liver fibrosis, and even hepatocellular carcinoma [4]. Recently, experts have proposed renaming NAFLD as “MAFLD” to better reflect its close association with metabolic disorders. According to global epidemiological surveys, the prevalence of MAFLD has reached 20%–30% in many countries, particularly in regions with high rates of obesity and diabetes [1,5]. Meanwhile, the global prevalence of CKD continues to rise, with a median prevalence of 9.5% (interquartile range 5.9%–11.7%). It is estimated that approximately 850 million people worldwide suffer from kidney disease, the majority of whom reside in low- and middle-income countries. With aging populations and population growth, the prevalence of CKD in low- and middle-income countries is expected to increase significantly in the coming decades. In contrast to cardiovascular diseases, stroke, and respiratory diseases, CKD mortality is on the rise, making it the third fastest-growing cause of death globally. By 2040, CKD is projected to be the fifth leading cause of years of life lost worldwide [6,7]. These conditions are closely linked to adverse outcomes, premature mortality, diminished quality of life, as well as substantial socioeconomic costs [8–10].
Epidemiological studies indicate a pronounced overlap in the incidence of MAFLD and CKD. Several prospective cohort studies have demonstrated that MAFLD independently increases the risk of renal impairment and CKD progression [11,12]. In addition, a large meta-analysis including over 1.2 million individuals showed that NAFLD nearly doubles the risk of developing CKD [13]. CT imaging analyses highlighted the presence of moderate-to-severe hepatic steatosis in a notable proportion of CKD patients [14]. A cohort study conducted in the United States reported a prevalence of MAFLD at 32.6%, with MAFLD patients being significantly older and exhibiting a higher incidence of CKD [15]. In contrast, a study targeting the middle-aged and elderly population in China found that the prevalence of MAFLD was as high as 46.7%, significantly exceeding the prevalence observed in other adult cohorts, with a 64% higher incidence of CKD among MAFLD patients [16]. This association not only underscores the close relationship between MAFLD and CKD, but also highlights the accelerated metabolic impairment in high-risk groups. Specifically, in the elderly population, MAFLD exacerbates the dual burden on both hepatic and renal functions, significantly increasing the risk of chronic diseases.
Furthermore, the burden of MAFLD and CKD demonstrates notable regional variations. For example, in North Africa and the Middle East, the prevalence of both MAFLD and CKD is significantly higher, which correlates with the metabolic risk factors and Socio-Demographic Index (SDI) of these regions. A data analysis based on the 2021 Global Burden of Disease Study revealed a significant association between high MAFLD prevalence and increased CKD incidence in countries with low SDI, whereas no such association was found in high SDI countries [17]. These findings emphasize the complex interplay between MAFLD and CKD, particularly in varying economic contexts, underscoring the urgency for targeted public health policies and interventions. As globalization progresses, understanding the epidemiological characteristics of different populations and regions is crucial for the formulation of effective public health strategies.
Mitochondria play a crucial role in cellular energy metabolism, influencing energy production, redox balance, and apoptosis [18–20]. Disruption of mitochondrial function not only affects hepatic lipid metabolism and detoxification but may also lead to inadequate energy supply in renal cells, exacerbating tubular damage and degeneration. Previous studies have identified mitochondrial dysfunction as a key factor in the pathogenesis of both MAFLD and CKD. In MAFLD, mitochondrial damage in the liver reduces fatty acid oxidation, promoting fat accumulation, increasing oxidative stress, and potentially leading to liver fibrosis and NASH [21]. In CKD patients, mitochondrial dysfunction intensifies oxidative stress and inflammatory responses and promotes renal fibrosis [22]. Therefore, mitochondrial damage may form a potential link between MAFLD and CKD, serving as a crucial intersection in the pathological progression of both diseases.
At present, there are no available treatments that specifically target both MAFLD and CKD, and the management of MAFLD is still primarily focused on symptom relief and prevention, with no therapeutic options addressing the underlying mechanisms of the disease. This article will explore mitochondrial dysfunction as a key interface between MAFLD and CKD and its role in the development of both diseases, providing insights that could help guide future treatment approaches for patients affected by these interconnected conditions.
2 Mitochondrial Vulnerability Shared by the Liver and Kidney
2.1 High Dependency of Liver and Kidney on Mitochondria
As the central hub of whole-body metabolism, the liver depends heavily on mitochondrial energy supply. Within hepatocytes, mitochondria not only generate adenosine triphosphate (ATP) but also directly participate in the metabolism of glucose, fatty acids, and amino acids. Through β-oxidation, the liver efficiently converts nutrient substrates into usable energy to sustain its diverse metabolic functions, thereby preserving lipid homeostasis and preventing excessive fat deposition [23,24]. Moreover, the liver regulates its metabolic functions by regulating a series of key signaling pathways. For example, when ATP consumption increases, the adenosine monophosphate-activated protein kinase (AMPK) pathway is activated, regulating intracellular metabolic balance and affecting glucose and lipid metabolism. When mitochondrial dysfunction occurs, oxidative stress and inflammatory signals activate pathways such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), contributing to the development of metabolic diseases like fatty liver and diabetes [23].
The kidney plays a pivotal role in maintaining water-electrolyte balance and acid-base homeostasis, making it one of the most energy-demanding organs [25]. Renal tissue is highly enriched with mitochondria, especially in tubular epithelial cells, where oxidative metabolism produces ATP to support essential ion transport mechanisms, such as the Na+, K+-ATPase pump [26]. These mechanisms ensure the effective regulation of sodium, potassium, calcium, and other ions by the kidney, thereby maintaining overall fluid and electrolyte balance [27]. In addition to ATP production, renal mitochondria are also involved in the regulation of cell death, calcium homeostasis, and steroid biosynthesis, underscoring their essential role in preserving cellular homeostasis [28–30].
2.2 Central Role of Mitochondrial Dysfunction in MAFLD and CKD
MAFLD develops in the context of metabolic abnormalities such as excessive energy intake, obesity, and insulin resistance, and is initially characterized by the accumulation of neutral lipids within hepatocytes. Excessive free fatty acids (FFAs) enter hepatocytes through membrane transport proteins, such as cluster of differentiation 36 (CD36). In the early stages, increased fatty acid uptake and triglyceride synthesis contribute to the development of simple steatosis [31]. However, as metabolic dysregulation persists, lipid overload precipitates lipotoxic effects, activating endoplasmic reticulum stress and oxidative stress pathways, thereby driving excessive production of reactive oxygen species (ROS). The combined impact of lipid overload and ROS accumulation disrupts mitochondrial structure and function, resulting in a decline in membrane potential, reduced efficiency of oxidative phosphorylation (OXPHOS), impaired electron transport chain (ETC) activity, and compromised ATP synthesis. In turn, mitochondrial dysfunction exacerbates ROS generation, forming a vicious cycle [32]. Moreover, oxidative damage to mitochondrial DNA limits its repair capacity, rendering hepatocytes more vulnerable to concurrent oxidative stress and inflammation, thereby facilitating the transition from simple steatosis to MAFLD and eventually to hepatic fibrosis [33].
CKD is defined as a decline in renal function persisting for more than three months, with an irreversible progressive course. This condition is often associated with metabolic disorders, particularly those linked to diabetes, hypertension, and obesity [34]. Although the kidney can initially compensate by adjusting glomerular filtration and renal blood flow to maintain basic function, ongoing metabolic disturbances exacerbate lipid and glucose dysregulation within renal cells, leading to the accumulation of metabolic by-products in renal tissue and triggering chronic low-grade inflammation and oxidative stress [35]. In the highly energy-dependent tubular epithelial cells, sustained ROS production results in a loss of mitochondrial membrane potential, impaired oxidative phosphorylation, and diminished ATP generation. Concurrently, impaired mitophagy prevents the removal of damaged mitochondria, further amplifying ROS accumulation and inflammatory activation. This vicious cycle progressively drives structural damage and functional decline in renal tissue, ultimately promoting the onset and progression of CKD [36].
3 Mitochondrial Dysfunction Mechanisms in MAFLD and CKD
3.1 ROS Overproduction and ETC Dysfunction
Mitochondria serve as the main source of ROS in cells. Under physiological conditions, about 0.2%–2% of electrons in the mitochondrial ETC undergo aberrant leakage, reacting with molecular oxygen to generate superoxide (O2−) and hydrogen peroxide (H2O2), rather than following the canonical oxidative phosphorylation pathway. Disruptions in ETC function or damage to the mitochondrial inner membrane significantly exacerbate this electron leakage phenomenon, resulting in excessive ROS generation [37].
To adapt to the oxygen-rich environment, aerobic organisms have developed sophisticated antioxidant defense mechanisms to maintain redox homeostasis by regulating intracellular ROS levels [38]. Superoxide dismutase (SOD), one critical component of this antioxidant system, demonstrates dynamic changes during disease progression. In the initial stages of MAFLD, the upregulation of SOD activity has been served as a part of the cellular stress response [38,39]. However, opposing evidence suggests a significant reduction in SOD activity in murine models fed a high-fat diet (HFD) [40]. This paradoxical reduction may be due to the exhaustion of antioxidant enzymes under sustained oxidative stress or the progressive hepatic injury. Consistently, kidney function correlates with marked attenuation of antioxidant defenses. Patients with CKD exhibit a weakened antioxidant defense system, with reduced SOD expression [41], and other crucial antioxidants, including catalase, glutathione peroxidase (GPx), intracellular glutathione (GSH), and thiol-containing compounds [42]. This systemic antioxidant deficiency likely contributes to the progression of uremic complications.
Any disruption in the ETC complexes can further increase ROS generation, with complexes I and III serving as the predominant sites of ROS production. Experimental evidence demonstrated that impaired ETC activity directly correlates with enhanced ROS generation. In a rodent model of MAFLD induced by 30-day choline-deficient dietary intervention, 35% reduction in complex I is concomitant with a significantly elevated H2O2 production in the isolated hepatic mitochondria [43]. Similarly, the 5/6 nephrectomy (5/6Nx) model, a well-established experimental model for CKD, revealed that sustained mitochondrial oxidative stress and uncoupling are mechanistically associated with an initial decline in complex I and ATP synthase activities, followed by a subsequent reduction in complex III activity [44].
3.2 Mitochondrial Membrane Potential (ΔΨm) Dysregulation
The mitochondrial membrane consists of the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM), each with distinct structures and functions. The OMM is porous, allowing the free diffusion of small molecules and ions, whereas the selective transport of larger molecules is mediated by specialized transport proteins. In contrast, the IMM served as a tight barrier that permits only specific ions and molecules to traverse via the highly selective transporters. The IMM is the principal site of oxidative phosphorylation, where the electrochemical gradient is generated and subsequently drives ATP synthesis. Due to its ion selectivity, the IMM maintains a transmembrane potential of approximately 180 mV, which is essential for mitochondrial bioenergetics [45].
Evidence suggests that mitochondrial membrane integrity is compromised in MAFLD due to the persistent opening of the mitochondrial permeability transition pore (MPTP) induced by excess FFAs. This pathological process is aggravated by mitochondrial Ca2+ overload and the dysregulation of key pore-forming components, such as adenine nucleotide translocase (ANT) and the F1F0-ATP synthase complex. Persistent MPTP activation disrupts mitochondrial ultrastructure, leading to matrix swelling, cristae disorganization, and IMM rupture. Moreover, increased mitochondrial membrane permeability facilitates the cytosolic release of cytochrome c, triggering the intrinsic apoptotic signaling cascade and accelerating the progression of MAFLD [46,47].
In experimental models of CKD, dysregulated IMM permeability has been identified as a critical mechanism underlying the loss of ΔΨm and subsequent cellular injury. Specifically, under conditions of heightened oxidative stress, abnormal MPTP opening markedly induces the nonselective IMM permeability, leading to ΔΨm collapse, mitochondrial structural disintegration, and ultimately tubular epithelial cell necrosis [48]. In addition, upregulation of uncoupling protein 2 (UCP-2) promotes proton leakage across the IMM, resulting in diminishing ΔΨm and increased oxygen consumption [49]. Similar phenomenon have been observed in diabetic kidney disease (DKD) models, where recurrent MPTP openings are strongly associated with tubular cell membrane depolarization, further supporting its putative role as a critical link between mitochondrial structural damage and renal functional impairment [50].
3.3 Mitochondrial Dynamics Imbalance
Mitochondria are highly dynamic organelles whose morphology and distribution are continuously remodeled through the processes of fusion and fission to meet the metabolic demands of the cell [51]. Mitochondrial fission primarily depends on dynamin-related protein 1 (DRP1) and its outer membrane receptor, fission protein 1 (FIS1). DRP1 assembles into ring-like structures on the mitochondrial surface and drives membrane constriction by hydrolyzing Guanosine Triphosphate (GTP), ultimately completing the division process. Fusion, on the other hand, is mediated by outer membrane proteins mitofusin 1 (MFN1) and mitofusin 2 (MFN2). Notably, MFN1 tethers opposing mitochondria through its HR2 domain, while MFN2 promotes mitochondrial fusion by oligomerizing with other MFN2 or MFN1 proteins. Additionally, the fusion of the inner membrane is regulated by optic atrophy 1 (OPA1) [52,53]. Excessive mitochondrial fusion can induce the formation of elongated tubular structures, while increased fission results in fragmented mitochondria [54]. These disruptions may cause oxidative stress and halt energy production, as well as trigger abnormal signaling pathways that contribute to numerous metabolic abnormalities. Maintaining a finely balanced interplay between these processes is essential for preserving mitochondrial integrity and cellular homeostasis, as any dysregulation ultimately leads to mitochondrial dysfunction and cellular damage.
In mice that were fed with an HFD, mitochondrial structural integrity was compromised across multiple organs, accompanied by marked downregulation of the fusion proteins MFN2 and OPA1, together with increased expression of the fission mediator DRP1 [55]. Similarly, exposure to FFAs has been shown to upregulate DRP1 and FIS1 while suppressing OPA1 and MFN2 expression [56]. From a mechanistic perspective, the deficiency of prohibitin-2 in the liver leads to an increase in the proteolytic cleavage of L-OPA1 [57]. Nevertheless, restoring L-OPA1 expression via adenovirus delivery reactivates mitochondrial function in hepatocytes and offers protection against NAFLD [57]. Using an HFD-induced NAFLD mouse model, it was observed that the degradation of OXPHOS subunits was elevated, resulting in mitochondrial dysfunction [58]. The underlying mechanism involves OXPHOS being the primary energy source for cells, driving ATP production and supplying the energy required for mitochondrial fusion. Furthermore, mitochondrial fusion is sensitive to OXPHOS activity, with the OXPHOS rate modulating the efficiency of inner mitochondrial membrane fusion, although its impact on the outer membrane remains minimal [59]. Furthermore, OXPHOS defects or inhibition can induce oxidative stress, which increases the production of ROS and further damages the respiratory chain [60].
Similar disruptions in mitochondrial dynamics have also been observed in CKD models. Two independent studies have shown that renal tubules from CKD models exhibited pronounced mitochondrial fragmentation [61,62]. In the 5/6Nx CKD model, isolated mitochondria displayed increased levels of FIS1 and DRP1, indicating excessive mitochondrial fission in CKD [63]. DRP1 is primarily localized in the cytoplasm, and upon activation, it translocates to the mitochondrial outer membrane through interaction with receptor proteins such as FIS1, where it exerts its function. The use of the DRP1 inhibitor P110 reduces mitochondrial translocation of DRP1 and disrupts the interaction between DRP1 and FIS1, providing significant protection in the mouse renal ischemia-reperfusion injury (IRI) model [64]. HFD feeding resulted in reduced MFN2 and OPA1 levels in the kidney, along with an increase in DRP1 expression [65]. In diabetic rat models, the time-course mapping of mitochondrial dynamics revealed that, in the early phase, mitochondrial fusion and biogenesis in renal proximal tubule epithelial cells were increased as a compensatory mechanism to excessive mitochondrial fission, which was enhanced by hyperglycemia. Regrettably, as mitochondrial fission became predominant, mitochondrial fragmentation increased, antioxidant levels decreased, and mitochondrial bioenergetic function was impaired [66]. These findings suggest that metabolic conditions, such as diabetes and HFD, exacerbate renal damage by disrupting mitochondrial dynamics.
Autophagy is a cellular recycling process that selectively removes damaged or dysfunctional mitochondria to prevent the accumulation of harmful components and to maintain cellular homeostasis and physiological function [67]. Therefore, in the adaptive phase, mitophagy is more associated with metabolic adaptation and homeostasis. In contrast, improper regulation of mitophagy leads to cellular damage, involving the accumulation of ROS, and triggers liver-related diseases such as steatosis, fibrosis, and cancer. The PTEN-induced kinase 1/Parkin RBR E3 ubiquitin-protein ligase (PINK1/Parkin) pathway is considered the principal route for mitophagy under cellular stress conditions [68,69]. Recent studies have highlighted the roles of mammalian target of rapamycin (mTOR) and AMPK in regulating mitophagy. mTOR, as a central regulator of cell growth and metabolism, inhibits mitophagy under nutrient-rich conditions by suppressing autophagy-related proteins. In contrast, AMPK is activated under energy stress and promotes mitophagy by inhibiting mTOR and activating autophagy proteins like ULK1. The interplay between mTOR and AMPK is crucial for maintaining mitochondrial health, particularly under metabolic stress, and plays an important role in diseases such as MAFLD [70].
Dysregulated mitophagy compromises mitochondrial renewal and quality control, impairing oxidative phosphorylation and energy production, which in turn disrupts lipid metabolism and drives hepatic steatosis [71]. Defective mitophagy has been observed in both HFD-induced NAFLD mouse models and in vitro cultured cells treated with oleic acid (OA) or palmitic acid (PA). In mouse models of NAFLD induced by a HFD, the abundance of PINK1 and Parkin is markedly reduced relative to controls, indicating the relationship between suppression of mitophagy-related proteins and liver injury [72]. In addition, pharmacologically enhancing PINK1/Parkin-dependent mitophagy by the plant flavonoid quercetin has been reported to alleviate HFD-induced hepatic disorders in a 10-week feeding mouse model [73]. Under energy stress, AMPK activates mitophagy by inhibiting mTOR, a negative regulator of autophagy. Studies have shown that omentin-1, a protein that influences metabolism, can restore AMPK activity and promote autophagy by normalizing the AMPK/mTOR pathway [74]. Further research found that PA treatment significantly suppressed the fluorescence intensity of p-AMPK and Parkin in hepatocytes. However, MST1 knockdown activated the AMPK pathway, restored Parkin expression, and reversed the inhibition of mitophagy [75].
Similar mitophagy defects occur in the kidney. In cisplatin-induced nephrotoxicity, impaired PINK1/Parkin-mediated mitophagy leads to the accumulation of damaged mitochondria, promoting oxidative stress and fibrotic remodeling [76]. Furthermore, nuclear factor erythroid 2-related factor 2 (NRF2) is a key redox transcription factor that helps cells resist oxidative stress and protects mitochondria from damage [77]. Recent studies have demonstrated that activating NRF2 can enhance PINK1/Parkin-mediated mitophagy in obesity-related CKD, leading to a significant reduction in oxidative stress and mitochondrial depolarization in renal tubular cells [65]. mTOR has been linked to the proliferation of tubular cells in DKD. However, when the mTOR gene is deleted, tubular damage is improved [78]. The mTORC1 inhibitor rapamycin exerts a critical therapeutic effect in CKD, potentially modulating disease progression through the regulation of the endogenous cannabinoid system [79]. Mitochondrial dysfunction emerges as a central mediator in the liver-kidney crosstalk of MAFLD and CKD, associated with defective mitophagy, imbalanced mitochondrial dynamics, ΔΨm collapse, and ROS-ETC dysfunction. These interconnected mechanisms create a vicious cycle of bioenergetic failure and cellular damage in both organs (Fig. 1).
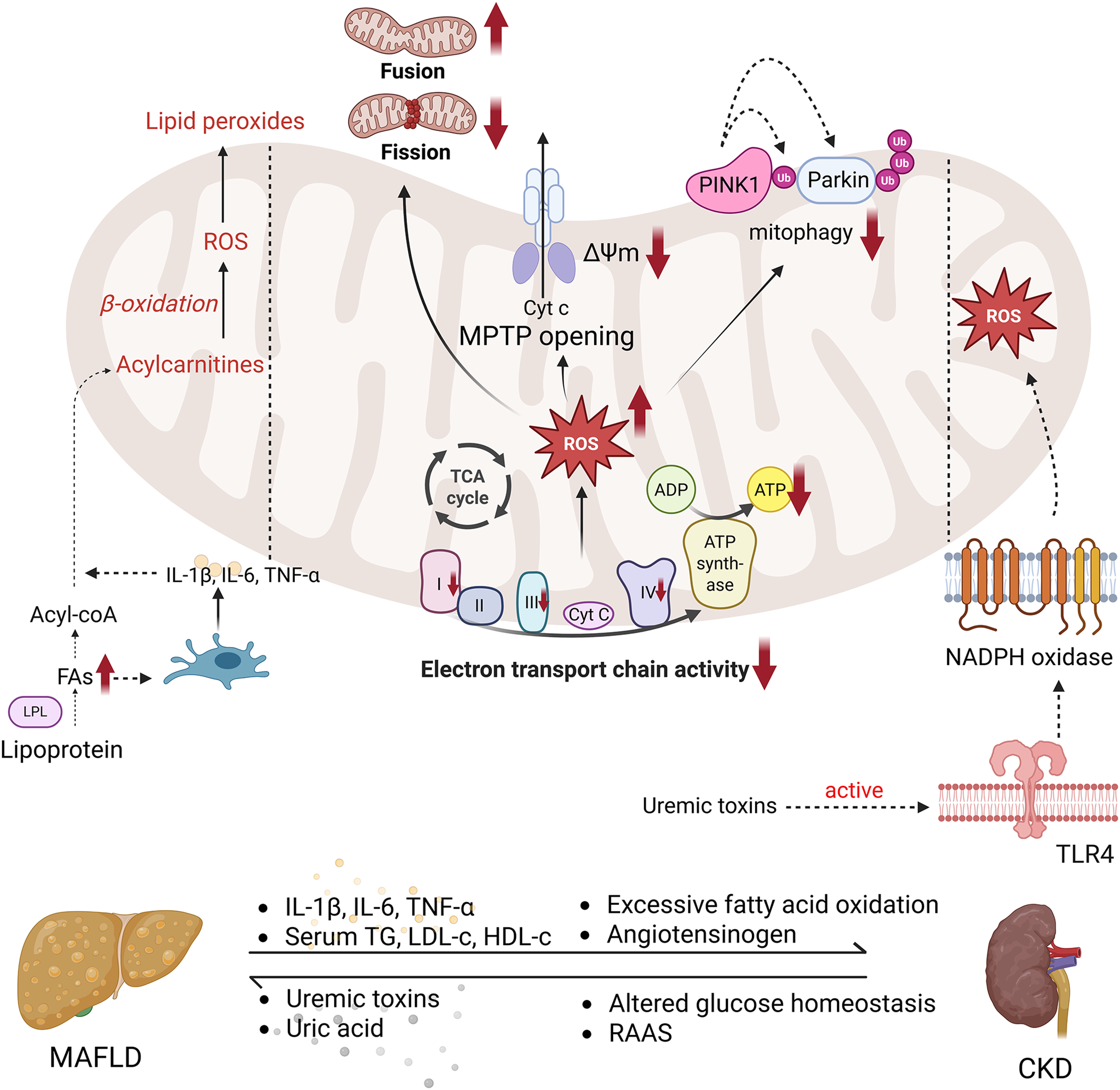
Figure 1: Mitochondrial dysfunction in MAFLD and CKD. In MAFLD, lipid overload leads to the accumulation of excessive FAs, which, through the action of LPL, stimulate the release of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α. This inflammatory response enhances mitochondrial β-oxidation, further exacerbating the accumulation of ROS. In CKD, the accumulation of uremic toxins activates TLR4 receptors, triggering NADPH oxidase activation and further promoting ROS generation, contributing to kidney injury. The shared mitochondrial dysfunction in both MAFLD and CKD is characterized by impaired ETC function, leading to excessive ROS production, reduced ATP synthase activity, and diminished ATP generation. This results in the loss of ΔΨm, MPTP opening, and Cyt c release, all of which drive apoptosis. Additionally, defective PINK1/Parkin-mediated mitophagy further disrupts mitochondrial quality control, while mitochondrial dynamics become imbalanced, marked by decreased fusion and increased fission. Abbreviation: TCA cycle (Tricarboxylic Acid Cycle), TLR4 (Toll-Like Receptor 4), LDL (Low Density Lipoprotein), HDL (High Density Lipoprotein), MAFLD (Metabolic Dysfunction-associated Fatty Liver Disease), CKD (Chronic Kidney Disease), FAs (Fatty Acids), LPL (Lipoprotein Lipase), IL-1β (Interleukin-1 beta), IL-6 (Interleukin-6), TNF-α (Tumor Necrosis Factor alpha), ROS (Reactive Oxygen Species), ETC (Electron Transport Chain), ATP (Adenosine Triphosphate), NADPH (Nicotinamide Adenine Dinucleotide Phosphate), ΔΨm (Mitochondrial Membrane Potential), MPTP (Mitochondrial Permeability Transition Pore), Cyt c (Cytochrome c), PINK1 (PTEN-induced kinase 1), and Parkin (Parkin RBR E3 Ubiquitin Protein Ligase), TG (Triglycerides), ADP (Adenosine Diphosphate), RAAS (renin–angiotensin–aldosterone system), Acyl-CoA (Acyl Coenzyme A). Created with Biorender
4 Mitochondrial Dysfunction and Liver–Kidney Crosstalk
4.1 Hepatic Lipid Overload Induces Renal Mitochondrial Injury
In recent years, accumulating epidemiological evidence demonstrates that MAFLD is not only a hepatic metabolic disorder but also an independent risk factor for CKD. A meta-analysis revealed that patients with MAFLD exhibit a roughly 35% higher prevalence of CKD and a 29% increased risk of new-onset CKD compared with those without MAFLD [80]. Notably, a retrospective cohort study identified abdominal obesity as a significant mediator, with MAFLD and abdominal obesity synergistically elevating both CKD prevalence and all-cause mortality [15]. These observations implicate dysregulated lipid accumulation as a potential pathological link between MAFLD and CKD.
In MAFLD, persistent hepatic fatty acid overload leads to excessive triglycerides (TGs) storage within lipid droplets. Once hepatic storage capacity is exceeded, TGs are assembled into very-low-density lipoproteins (VLDLs) and secreted into the circulation [81]. These ectopic lipid deposition in non-adipose tissues, including the kidneys, disrupt local metabolism and induce cellular damage [82]. The uptake of FFAs in the kidneys is mediated by key transporters, including CD36 fatty acid transport proteins (FATPs), and fatty acid binding proteins (FABPs) [83]. These transporters facilitate the entry of FFAs across the plasma membrane into the cytoplasm, where they are either utilized for energy production or stored as triglycerides. CD36, a prominent FFA transporter, plays a crucial role in this process and is widely expressed in various renal cell types, including tubular epithelial cells, podocytes, and mesangial cells [84]. The overexpression of CD36 in renal cells can play a key role in promoting lipid accumulation in the tubules, impairing mitochondrial function, and activating inflammatory pathways [85,86].
4.2 Uremic Toxins Promote Hepatic Mitochondrial Injury
Clinical studies underscore the bidirectional liver-kidney axis in advanced CKD. A prospective study reported that 56% patients with severe renal impairment, particularly those hemodialysis-dependent ones, were diagnosed with NAFLD using a combination of controlled attenuation parameter (CAP) and ultrasound, highlighting the interplay between renal dysfunction and hepatic steatosis [87]. Similarly, among end-stage renal disease (ESRD) patients with type 2 diabetes (T2D), ultrasound-diagnosed NAFLD was present in 69.2% [88]. Such high comorbidity suggests that the uremic toxins, through the circulation in advanced CKD, may systemically exacerbate organ injury, where the liver being a primary target [89]. Indoxyl sulfate (IS) is a protein-bound uremic toxin (PBUT), primarily bound to serum proteins such as albumin [90]. Due to the large size of its complex, it cannot be effectively cleared by the dialysis membrane pores, and this retention can lead to liver involvement [91]. Additionally, IS activates AhR, leading to upregulation of hepatic P-glycoprotein (P-gp) expression and activity, further affecting its transport and hepatic detoxification functions [91]. IS also affects the liver through the gut-liver axis by inhibiting the expression of selenoprotein P (SEPP1), causing selenium deficiency, which subsequently induces hepatic ferroptosis [92]. Furthermore, as a metabolic product of gut microbiota, IS is converted into indoxyl sulfate in the liver via cytochrome P450 enzymes, such as CYP2E1, a process that intensifies oxidative stress in the liver [93].
4.3 Damage-Associated Mitochondrial Products
Excessive ROS generation is a hallmark of mitochondrial dysfunction in MAFLD and CKD. Beyond comprising mitochondrial integrity and bioenergetics, ROS simultaneously act as signaling mediators that drive immune-inflammatory cascades, ultimately exacerbating hepatic and renal injury [19]. Mechanistically, ROS induce MPTP opening, facilitating the cytoplasmic release of mitochondrial DNA (mtDNA). As a canonical damage-associated molecular pattern (DAMP), mtDNA is recognized by host cell sensors, activating the cGAS/STING pathway and the NLRP3 inflammasome [94–96], thereby triggering the production of type I interferons (IFN-I) and interleukin-1β (IL-1β) [97]. Concurrently, ROS activate pro-inflammatory signaling cascades, upregulating cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) [98]. These circulating cytokines propagate along the liver–kidney axis and amplify systemic inflammatory responses and multi-organ dysfunction [99,100].
In CKD, the renin–angiotensin–aldosterone system (RAAS) remains chronically overactivated, with elevated angiotensin II (Ang II) levels [101]. In NAFLD, sustained hepatic inflammation and oxidative stress likewise stimulate hepatic angiotensinogen production [102], thereby exacerbating RAAS activation. Such dysregulation of RAAS is not organ-restricted; systemic RAAS activation exacerbates hepatocellular injury [103], thereby establishing a pathological vicious cycle between the liver and kidneys.
Elevated Ang II levels contribute to vascular dysfunction, exacerbating ischemic injury in both the kidney and liver. In the kidney, Ang II induces vasoconstriction of glomerular capillary and endothelial dysfunction, ultimately leading to increased permeability of the filtration barrier and production of proteinuria [104]. In the liver, Ang II elevates intrahepatic vascular resistance, thereby exacerbating hepatocellular ischemia and fostering fibrogenesis [105]. At the cellular level, Ang II activates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and impairs mitochondrial complexes I and III, resulting in excessive mtROS production and mitochondrial dysfunction. These mechanisms accelerate the progression of MAFLD and CKD [106–108]. The interplay between hemodynamic disturbances and mitochondrial stress amplifies pathological liver–kidney crosstalk, implicating the critical role of RAAS dysregulation in the progression of comorbidity conditions.
Emerging evidence underscores that mitochondrial dysfunction serves as a critical nexus in the bidirectional crosstalk between MAFLD and CKD. Dysregulated RAAS activation, damage-associated mitochondrial products, uremic toxin accumulation, and hepatic lipid overload synergistically exacerbate both hepatic and renal mitochondrial injury, perpetuating a self-amplifying cycle of metabolic and inflammatory damage. Therapeutic strategies targeting mitochondrial protection or interrupting these organ-specific insults may offer novel approaches to mitigate multi-organ dysfunction in this high-risk population (Fig. 2).
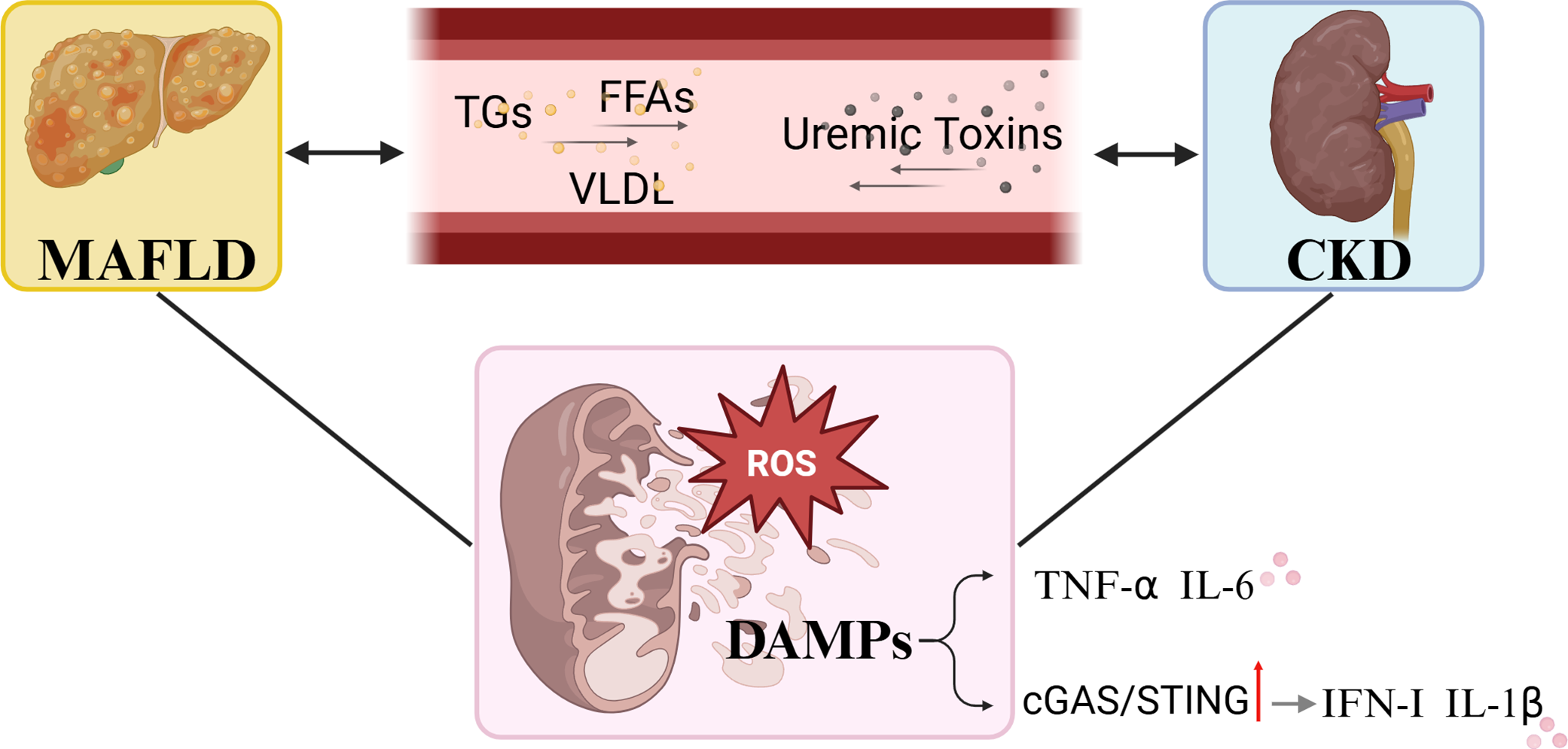
Figure 2: Crosstalk between MAFLD and CKD. MAFLD can lead to lipid overload, promoting the onset and progression of CKD through the bloodstream, while uremic toxins in CKD can circulate and worsen the course of MAFLD, collectively causing mitochondrial oxidative stress injury. Excessive ROS induces the release of DAMPs, which in turn activate pro-inflammatory cytokines. cGAS (cyclic GMP-AMP Synthase), STING (Stimulator of Interferon Genes), MAFLD (Metabolic Dysfunction-associated Fatty Liver Disease), CKD (Chronic Kidney Disease), ROS (Reactive Oxygen Species), DAMPs (Damage-Associated Molecular Patterns), TNF-α (Tumor Necrosis Factor alpha), IL-6 (Interleukin-6), IFN-I (Interferon type I), IL-1β (Interleukin-1 beta). TG (Triglycerides), FFAs (Free Fatty Acids), VLDL (Very Low-Density Lipoprotein). Created with Biorender
5 Clinical Relevance of Mitochondrial Dysfunction in MAFLD and CKD
5.1 Mitochondrial Dysfunction across Disease Stages
In MAFLD, mitochondrial dysfunction begins to manifest early in the disease process. Initially, lipid accumulation within hepatocytes results in mild mitochondrial impairment, characterized by reduced ATP synthesis and subtle oxidative stress. At this stage, mitochondria can adapt to environmental changes through proliferation [109]. However, as the disease progresses to NASH and liver fibrosis, the inflammation and fat accumulation in the liver intensify, leading to a significant decline in mitochondrial oxidative phosphorylation function. This results in an increase in ROS and exacerbates cellular damage, ultimately leading to irreversible apoptosis [110].
Compared to MAFLD, mitochondrial dysfunction in CKD is primarily associated with tubular damage and apoptosis. Early in CKD, mitochondrial impairment manifests as reduced ATP synthesis and elevated ROS levels. Given the high metabolic demands of renal tubular cells, these changes rapidly compromise kidney function [111,112]. As the disease progresses, a decline in mitochondrial membrane potential and exacerbated oxidative stress lead to tubular cell death, which in turn accelerates kidney fibrosis and functional decline. The combined effects of metabolic dysregulation and inflammation in CKD result in mitochondrial dysfunction that is more pronounced than in MAFLD, contributing to a more complex pathophysiological progression [112,113]. Clinically, monitoring mitochondrial biomarkers can help in assessing disease stage and progression, providing valuable insights for early diagnosis and potential therapeutic interventions.
5.2 Mitochondrial Biomarkers in Diagnosis
Given the critical role of mitochondria in liver and kidney diseases, clinicians can assess mitochondrial function and injury through biomarkers measured in liver or kidney biopsy specimens. Changes in mtDNA integrity and copy number serve as indicators of mitochondrial function, with alterations often linked to mitochondrial dysfunction and various pathological conditions [114]. Furthermore, the expression of mitochondrial fusion and fission proteins, such as MFN2 and DRP1, reflects shifts in mitochondrial dynamics, with frequent dysregulation observed in liver and kidney diseases [115]. Similarly, the expression of mitochondrial biogenesis-related proteins, such as PGC-1α [116], serves as a marker of mitochondrial proliferation and functional recovery. Moreover, oxidative stress markers, including ROS and byproducts like 8-hydroxy-2-deoxyguanosine, indicate oxidative damage caused by excessive mitochondrial activity, shedding light on the extent of mitochondrial injury [117]. In addition, cytochrome c, essential for mitochondrial function and apoptosis, is released into the cytosol following mitochondrial damage, thereby initiating the apoptotic pathway [118]. Another critical protein complex, mitochondrial ATP synthase (comprising ATP5A and ATP5B), is responsible for ATP synthesis in the inner mitochondrial membrane [119]. Dysfunction of ATP synthase is closely associated with metabolic disturbances, particularly in energy-demanding organs such as the liver and kidneys. We can assess mitochondrial dysfunction in the liver and kidneys by measuring these biomarkers, which can guide decisions regarding the potential use of mitochondria-targeted therapies.
5.3 Mitochondria-Targeted Therapies
MAFLD and CKD share overlapping pathogenic mechanisms, with mitochondrial dysfunction emerging a critical therapeutic target. Recent advances have identified several mitochondria-directed strategies that demonstrate protective effects against both hepatic and renal injury in preclinical and clinical studies.
MitoQ, a mitochondria-targeted derivative of coenzyme Q10, selectively accumulates in the IMM in a membrane potential-dependent manner. It serves as a mitochondrial-targeted antioxidant that stabilizes mitochondrial membrane potential, mitigates mitochondrial damage induced by membrane potential loss, and reduces the generation of ROS [120]. MitoQ also activates AMPK and its downstream signaling pathways, regulating autophagy, redox homeostasis, and inflammatory responses, ultimately contributing to liver and kidney protection [121,122]. A study in HFD-fed rats found that MitoQ alleviated hepatic steatosis, possibly through upregulation of mitochondrial cardiolipin and subsequent enhancement of respiratory chain complexes II, III, and V [123]. Furthermore, a randomized controlled trial in patients with stage 3–5 CKD revealed that four weeks of daily MitoQ supplementation significantly improved macrovascular and microvascular function, notably enhancing endothelial-dependent vasodilation and microcirculatory perfusion, compared to placebo [124].
Szeto–Schiller (SS) peptides, including the lead compound SS-31 (elamipretide) represent a novel class of mitochondria-targeted protective agents which bind to cardiolipin, thereby stabilizing the IMM, enhancing bioenergetic efficiency, and suppressing mtROS generation [125,126]. In murine models of CKD, elamipretide attenuates CKD progression by restoring mitochondrial membrane integrity, attenuating oxidative damage, and inhibiting fibrotic remodeling [127]. Similarly, in murine models of type 2 diabetes, SS-31 preserved hepatic mitochondrial function by improving H2O2 detoxification, mitigating lipid peroxidation, and sustaining ATP production [128].
Cyclosporine A (CsA) is a widely used immunosuppressant that inhibits the opening of the MPTP, thereby protecting mitochondrial integrity. By binding to cyclophilin D, CsA blocks its interaction with adenine nucleotide translocator (ANT), preventing the opening of the MPTP [129]. In a doxorubicin-induced kidney injury rat model, CsA significantly inhibited the release of cytochrome c (Cyt c) and cell apoptosis, improving mitochondrial function [130]. Additionally, as a potent MPTP inhibitor, CsA alleviates mitochondrial damage induced by FFAs, mitigates the opening of the MPTP, and reduces hepatic steatosis and oxidative stress in mice [47]. In addition to the well-known MPTP-targeting drug CsA, several novel compounds are also making innovative strides in MPTP-targeted therapies. For example, Phloretin suppresses the abnormal opening of the MPTP via the SH2 domain-containing protein tyrosine phosphatase 2/Janus kinase 2/BCL2 associated X, apoptosis regulator (SHP-2/JAK2/BAX) axis, thereby ameliorating NASH [131]. Another novel drug is Mn3O4 nanoparticles (C-Mn3O4 NPs), which preserve mitochondrial integrity by inhibiting MPTP activation and maintaining cellular redox balance in a cisplatin-induced CKD model [132]. Although many MPTP-targeting drugs are currently under development, they remain at an early stage, with limited further studies to confirm their efficacy, and none have entered clinical trials.
Accumulating evidence suggests that melatonin, beyond its antioxidant role, exerts broad protective effects by modulating mitochondrial dynamics and energy metabolism. In ob/ob mice, melatonin restores the expression of respiratory chain complexes II and V, enhanced oxygen phosphorylation, and maintains mitochondrial dynamics via balanced MFN2/DRP1 regulation [133]. Clinical data further support these findings: a randomized, double-blind trial in NAFLD patients demonstrated that the 6 mg/day melatonin supplement for 12 weeks significantly improved liver enzyme level, inflammation, and steatosis [134]. Moreover, in Zucker diabetic fatty rats, long-term melatonin administration preserved renal mitochondrial dynamics by promoting fusion protein expression, suppressing elevation of fission proteins, and concomitantly reducing ROS accumulation [135].
Originally developed for diabetes management, sodium–glucose cotransporter 2 (SGLT2) inhibitors (e.g., dapagliflozin, empagliflozin, canagliflozin) exhibit broad benefits in MAFLD and CKD. Mechanistically, these SGLT2 inhibitors activate the AMPK–PGC-1α pathway, thereby inducing PINK1/Parkin mediated mitophagy, thereby reducing oxidative stress and inflammatory in proximal tubules [136]. The DAPA-CKD trial further confirmed that SGLT2 inhibitors significantly improved clinical outcomes across diverse CKD populations [137]. In patients with type 2 diabetes and NAFLD, empagliflozin treatment significantly reduces hepatic fat content and liver function [138]. Moreover, empagliflozin also enhances hepatic autophagic flux, attenuates hepatic inflammation, lipid accumulation, and early fibrosis in the models of metabolic liver disease [139].
Nanotechnology has rapidly advanced in recent years, offering significant benefits in healthcare and related fields. Nanoparticles have shown great therapeutic potential in drug delivery, combating life-threatening diseases, tissue engineering, and other areas. Recent studies have demonstrated that temperature-responsive nanocarriers can effectively prevent the efflux of doxorubicin (DOX) and promote its accumulation in DOX-resistant tumors, as well as mitochondrial targeting [140]. Apart from cancer, recent research has also focused on targeted drug delivery for kidney injury. By combining cerium oxide nanoparticles (CeO2) with atorvastatin, this system can more effectively target mitochondria and eliminate ROS. In vitro experiments indicate that this drug delivery system can efficiently release the drug and specifically target mitochondria to clear excessive ROS, thereby reducing oxidative stress [141]. Cerium oxide nanoparticles not only serve as drug delivery carriers but also emerge as a novel and potent antioxidant with therapeutic properties, particularly in experimental liver diseases. CeO2 nanoparticles have been reported to act as scavengers for ROS and reactive nitrogen species (RNS), and exhibit multi-enzyme mimetic activities, such as SOD-like activity [142].
The substantial overlap in mitochondrial dysfunction between MAFLD and CKD underscores the need for therapies targeting inter-organ crosstalk. Table 1 summarizes the drugs and their mechanisms. Currently, the CKD management has transitioned from traditional symptomatic support to a mechanism-based strategies (e.g., SGLT2 inhibitors). In contrast, MAFLD treatment remains nascent, with lifestyle interventions as the cornerstone [143]. The disparity in therapeutic progress further underscores the urgency for developing cross-adaptable therapeutics to address this prevalent comorbidity.

5.4 Prevention Strategies for High-Risk Populations
The prevention strategies for high-risk populations should focus on early identification and intervention. For individuals with a family history of disease, it is recommended to combine routine clinical tests (such as liver and kidney function, blood lipids, and blood glucose levels). When necessary, liver and kidney biopsies should be performed to obtain mitochondrial biomarkers (e.g., mtDNA integrity, mitochondrial fusion and fission proteins MFN2, DRP1, oxidative stress markers such as ROS and their byproducts) for the timely identification of potential mitochondrial dysfunction. For individuals already diagnosed with a condition, timely treatment and stabilization of the disease are crucial. For example, MAFLD patients should aim for weight loss and fat reduction to prevent further hepatic fat accumulation, which not only improves liver metabolism but also enhances mitochondrial function and reduces oxidative stress. For patients with CKD, particularly those with severe hepatic dysfunction, standard pharmacological treatment and dialysis can effectively remove uremic toxins, alleviating liver damage and preventing further mitochondrial injury. Although mitochondrial-targeted therapies are not widely used yet, existing drugs like SGLT2 inhibitors have shown effectiveness in CKD and metabolic diseases by improving mitochondrial function and reducing oxidative stress, helping to control disease progression. Therefore, the recommendations for high-risk populations should focus on early identification and intervention of underlying causes, along with standardized, targeted treatment.
MASLD and CKD are escalating worldwide, placing a substantial public health burden. Although their distinct organ-specific manifestations, these two conditions exhibit bidirectional pathological interplay, mutually exacerbating disease progression through overlapping molecular mechanisms. Central to their pathogenesis are mitochondrial dysfunction, oxidative stress, and related pathological processes which collectively exacerbating disease progression. Disruption of the ETC leads to ROS generation, intensifying oxidative stress and further impairing mitochondrial integrity. Excessive ROS not only amplifies oxidative damage to cellular components but also triggers inflammatory pathways, thereby contributing to both hepatic and renal tissue injury. Additionally, the loss of mitochondrial membrane potential, coupled with the persistent opening of the MPTP, disrupts ATP synthesis and precipitates cellular damage.
Dysregulated mitochondrial dynamics, characterized by imbalances between mitochondrial fusion and fission, promotes mitochondrial fragmentation and functional impairment. Furthermore, impaired mitophagy due to the PINK1/Parkin pathway, leads to the defective clearance of damaged mitochondria, perpetuating oxidative stress and fosters fibrotic remodeling. These interconnected mitochondrial disturbances constitute critical pathogenic linking between MAFLD and CKD pathogenesis, offering potential therapeutic avenues for intervention.
7 Limitations and Future Perspectives
While Mitochondrial dysfunction has emerged as a promising therapeutic target in MAFLD and CKD, current strategies remain predominantly experimental, with limited clinical validation. Preclinical evidence suggests that interventions targeting oxidative stress, membrane stability, mitophagy, and mitochondrial dynamics may restore mitochondrial homeostasis, yet their clinical translation remains challenging. Future research needs to elucidate stage-dependent mitochondrial alterations in MAFLD and CKD progression, identify druggable targets within mitochondrial pathways, and rigorously evaluate the efficacy and safety of strategies to restore mitochondrial function. In this context, mitochondria represent not only vulnerable organelles but also a pivotal point, offering a promising platform for integrated interventions targeting liver-kidney axis dysfunction.
Acknowledgement: None.
Funding Statement: This work was supported by Top Talent Support Program for young and middle-aged people of Wuxi Health Committee (Grant No. HB2023008), Top Medical Expert Team of Wuxi Taihu Talent Plan (Grant Nos. DJTD202106, GDTD202105 and YXTD202101), Medical Key Discipline Program of Wuxi Health. Commission (Grant Nos. ZDXK2021007 and CXTD2021005), Top Talent Support Program for Young and Middle-Aged People of Wuxi Health Committee (Grant No. BJ2023090), and Scientific Research Program of Wuxi Health Commission (Grant Nos. Z20210 and M202208).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Jia Xia, Yutong Hou; literature collection and summary: Congwei You, Anwen Yin; draft manuscript preparation: Congwei You, Anwen Yin; writing—review & editing: Congwei You, Jia Xia, Yutong Hou; supervision: Le Zhang, Xiaolei Wang. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Guo Z, Wu D, Mao R, Yao Z, Wu Q, Lv W. Global burden of MAFLD, MAFLD related cirrhosis and MASH related liver cancer from 1990 to 2021. Sci Rep. 2025;15(1):7083. doi:10.1038/s41598-025-91312-5. [Google Scholar] [PubMed] [CrossRef]
2. Deng L, Guo S, Liu Y, Zhou Y, Liu Y, Zheng X, et al. Global, regional, and national burden of chronic kidney disease and its underlying etiologies from 1990 to 2021: a systematic analysis for the Global Burden of Disease Study 2021. BMC Public Health. 2025;25(1):636. doi:10.1186/s12889-025-21851-z. [Google Scholar] [PubMed] [CrossRef]
3. Liu Q, Zhao G, Li Q, Wu W, Zhang Y, Bian H. A comparison of NAFLD and MAFLD diagnostic criteria in contemporary urban healthy adults in China: a cross-sectional study. BMC Gastroenterol. 2022;22(1):471. doi:10.1186/s12876-022-02576-4. [Google Scholar] [PubMed] [CrossRef]
4. Parola M, Pinzani M. Liver fibrosis in NAFLD/NASH: from pathophysiology towards diagnostic and therapeutic strategies. Mol Aspects Med. 2024;95:101231. doi:10.1016/j.mam.2023.101231. [Google Scholar] [PubMed] [CrossRef]
5. Shi A, Deng J, Ma J, Yang L, Tantai X, Wang Q, et al. Prevalence and risk factors of metabolic dysfunction-associated fatty liver disease with renal insufficiency in overweight/obese adults. Obes Facts. 2023;16(6):548–58. doi:10.1159/000533626. [Google Scholar] [PubMed] [CrossRef]
6. Francis A, Harhay MN, Ong ACM, Tummalapalli SL, Ortiz A, Fogo AB, et al. Chronic kidney disease and the global public health agenda: an international consensus. Nat Rev Nephrol. 2024;20(7):473–85. doi:10.1038/s41581-024-00820-6. [Google Scholar] [PubMed] [CrossRef]
7. Bello AK, Okpechi IG, Levin A, Ye F, Damster S, Arruebo S, et al. An update on the global disparities in kidney disease burden and care across world countries and regions. Lancet Glob Health. 2024;12(3):e382–95. [Google Scholar] [PubMed]
8. Wong VW, Ekstedt M, Wong GL, Hagström H. Changing epidemiology, global trends and implications for outcomes of NAFLD. J Hepatol. 2023;79(3):842–52. doi:10.1016/j.jhep.2023.04.036. [Google Scholar] [PubMed] [CrossRef]
9. Karlsen TH, Sheron N, Zelber-Sagi S, Carrieri P, Dusheiko G, Bugianesi E, et al. The EASL-lancet liver commission: protecting the next generation of Europeans against liver disease complications and premature mortality. Lancet. 2022;399(10319):61–116. doi:10.1016/S0140-6736(21)01701-3. [Google Scholar] [PubMed] [CrossRef]
10. Bilson J, Mantovani A, Byrne CD, Targher G. Steatotic liver disease, MASLD and risk of chronic kidney disease. Diabetes Metab. 2024;50(1):101506. doi:10.1016/j.diabet.2023.101506. [Google Scholar] [PubMed] [CrossRef]
11. Heo JH, Lee MY, Kim SH, Zheng MH, Byrne CD, Targher G, et al. Comparative associations of non-alcoholic fatty liver disease and metabolic dysfunction-associated steatotic liver disease with risk of incident chronic kidney disease: a cohort study. Hepatobiliary Surg Nutr. 2024;13(5):801–13. doi:10.21037/hbsn-23-558. [Google Scholar] [PubMed] [CrossRef]
12. Gao J, Li Y, Zhang Y, Zhan X, Tian X, Li J, et al. Severity and remission of metabolic dysfunction-associated fatty/steatotic liver disease with chronic kidney disease occurrence. J Am Heart Assoc. 2024;13(5):e032604. doi:10.1161/JAHA.123.032604. [Google Scholar] [PubMed] [CrossRef]
13. Mantovani A, Petracca G, Beatrice G, Csermely A, Lonardo A, Schattenberg JM, et al. Non-alcoholic fatty liver disease and risk of incident chronic kidney disease: an updated meta-analysis. Gut. 2022;71(1):156–62. doi:10.1136/gutjnl-2020-323082. [Google Scholar] [PubMed] [CrossRef]
14. Adrian T, Sørensen IMH, Knop FK, Bro S, Ballegaard ELF, Nordestgaard BG, et al. Prevalence of non-alcoholic fatty liver disease in patients with chronic kidney disease: a cross-sectional study. Nephrol Dial Transplant. 2022;37(10):1927–34. doi:10.1093/ndt/gfab266. [Google Scholar] [PubMed] [CrossRef]
15. Cen C, Fan Z, Ding X, Tu X, Liu Y. Associations between metabolic dysfunction-associated fatty liver disease, chronic kidney disease, and abdominal obesity: a national retrospective cohort study. Sci Rep. 2024;14(1):12645. doi:10.1038/s41598-024-63386-0. [Google Scholar] [PubMed] [CrossRef]
16. Liang Y, Chen H, Liu Y, Hou X, Wei L, Bao Y, et al. Association of MAFLD with diabetes, chronic kidney disease, and cardiovascular disease: a 4.6-year cohort study in China. J Clin Endocrinol Metab. 2022;107(1):88–97. doi:10.1210/clinem/dgab641. [Google Scholar] [PubMed] [CrossRef]
17. Bai J, Zhang L, Zhang M, Hao Y, Yi Z, Zhou Y. Regional insights into the relationship between metabolic associated steatotic liver disease and chronic kidney disease: a socioeconomic perspective on disease correlation. BMC Public Health. 2025;25(1):993. doi:10.1186/s12889-025-22188-3. [Google Scholar] [PubMed] [CrossRef]
18. Zong Y, Li H, Liao P, Chen L, Pan Y, Zheng Y, et al. Mitochondrial dysfunction: mechanisms and advances in therapy. Sig Transduct Target Ther. 2024;9:124. doi:10.1038/s41392-024-01839-8. [Google Scholar] [PubMed] [CrossRef]
19. Sun A, Pollock CA, Huang C. Mitochondria-targeting therapeutic strategies for chronic kidney disease. Biochem Pharmacol. 2025;231:116669. doi:10.1016/j.bcp.2024.116669. [Google Scholar] [PubMed] [CrossRef]
20. Radosavljevic T, Brankovic M, Samardzic J, Djuretić J, Vukicevic D, Vucevic D, et al. Altered mitochondrial function in MASLD: key features and promising therapeutic approaches. Antioxidants. 2024;13(8):906. doi:10.3390/antiox13080906. [Google Scholar] [PubMed] [CrossRef]
21. Prasun P, Ginevic I, Oishi K. Mitochondrial dysfunction in nonalcoholic fatty liver disease and alcohol related liver disease. Transl Gastroenterol Hepatol. 2021;6:4. doi:10.21037/tgh-20-125. [Google Scholar] [PubMed] [CrossRef]
22. Pavlović N, Križanac M, Kumrić M, Vukojević K, Božić J. Mitochondrial dysfunction: the silent catalyst of kidney disease progression. Cells. 2025;14(11):794. doi:10.3390/cells14110794. [Google Scholar] [PubMed] [CrossRef]
23. Morio B, Panthu B, Bassot A, Rieusset J. Role of mitochondria in liver metabolic health and diseases. Cell Calcium. 2021;94:102336. doi:10.1016/j.ceca.2020.102336. [Google Scholar] [PubMed] [CrossRef]
24. Dabravolski SA, Bezsonov EE, Baig MS, Popkova TV, Orekhov AN. Mitochondrial lipid homeostasis at the crossroads of liver and heart diseases. Int J Mol Sci. 2021;22(13):6949. doi:10.3390/ijms22136949. [Google Scholar] [PubMed] [CrossRef]
25. Korus J, Szymczak M, Gołębiowski M, Rydzek J, Majcherczyk K, Wilk J, et al. Metabolic acidosis in patients with chronic kidney disease: diagnosis, pathogenesis, and treatment-a narrative review. Diagnostics. 2025;15(16):2052. doi:10.3390/diagnostics15162052. [Google Scholar] [PubMed] [CrossRef]
26. Wang XL, Li L, Meng X. Interplay between the redox system and renal tubular transport. Antioxidants. 2024;13(10):1156. doi:10.3390/antiox13101156. [Google Scholar] [PubMed] [CrossRef]
27. Cheng CJ, Nizar JM, Dai DF, Huang CL. Transport activity regulates mitochondrial bioenergetics and biogenesis in renal tubules. FASEB J. 2024;38(10):e23703. doi:10.1096/fj.202400358rr. [Google Scholar] [PubMed] [CrossRef]
28. Lu M, Zhan Z, Li D, Chen H, Li A, Hu J, et al. Protective role of vitamin D receptor against mitochondrial calcium overload from PM2.5-Induced injury in renal tubular cells. Redox Biol. 2025;80:103518. doi:10.1016/j.redox.2025.103518. [Google Scholar] [PubMed] [CrossRef]
29. Gao Q, Shi K, Shi X, Liu Y, Zhang H, Weng Q. 1,25(OH)2D3 up-regulated mitochondrial dynamics and biogenesis to modulate steroidogenesis in the scent glands of muskrats (Ondatra zibethicus). J Steroid Biochem Mol Biol. 2025;252:106787. doi:10.1016/j.jsbmb.2025.106787. [Google Scholar] [PubMed] [CrossRef]
30. Liu F, Yang Z, Li J, Wu T, Li X, Zhao L, et al. Targeting programmed cell death in diabetic kidney disease: from molecular mechanisms to pharmacotherapy. Mol Med. 2024;30(1):265. doi:10.1186/s10020-024-01020-5. [Google Scholar] [PubMed] [CrossRef]
31. Rada P, González-Rodríguez Á, García-Monzón C, Valverde ÁM. Understanding lipotoxicity in NAFLD pathogenesis: is CD36 a key driver? Cell Death Dis. 2020;11(9):802. doi:10.1038/s41419-020-03003-w. [Google Scholar] [PubMed] [CrossRef]
32. Nassir F. NAFLD: mechanisms, treatments, and biomarkers. Biomolecules. 2022;12(6):824. doi:10.3390/biom12060824. [Google Scholar] [PubMed] [CrossRef]
33. Mu C, Wang S, Wang Z, Tan J, Yin H, Wang Y, et al. Mechanisms and therapeutic targets of mitochondria in the progression of metabolic dysfunction-associated steatotic liver disease. Ann Hepatol. 2025;30(1):101774. doi:10.1016/j.aohep.2024.101774. [Google Scholar] [PubMed] [CrossRef]
34. Reiss AB, Jacob B, Zubair A, Srivastava A, Johnson M, De Leon J. Fibrosis in chronic kidney disease: pathophysiology and therapeutic targets. J Clin Med. 2024;13(7):1881. doi:10.3390/jcm13071881. [Google Scholar] [PubMed] [CrossRef]
35. Zhang X, Lerman LO. The metabolic syndrome and chronic kidney disease. Transl Res. 2017;183:14–25. doi:10.1016/j.trsl.2016.12.004. [Google Scholar] [PubMed] [CrossRef]
36. Kadatane SP, Satariano M, Massey M, Mongan K, Raina R. The role of inflammation in CKD. Cells. 2023;12(12):1581. doi:10.3390/cells12121581. [Google Scholar] [PubMed] [CrossRef]
37. Tirichen H, Yaigoub H, Xu W, Wu C, Li R, Li Y. Mitochondrial reactive oxygen species and their contribution in chronic kidney disease progression through oxidative stress. Front Physiol. 2021;12:627837. doi:10.3389/fphys.2021.627837. [Google Scholar] [PubMed] [CrossRef]
38. Jomova K, Alomar SY, Alwasel SH, Nepovimova E, Kuca K, Valko M. Several lines of antioxidant defense against oxidative stress: antioxidant enzymes, nanomaterials with multiple enzyme-mimicking activities, and low-molecular-weight antioxidants. Arch Toxicol. 2024;98(5):1323–67. doi:10.1007/s00204-024-03696-4. [Google Scholar] [PubMed] [CrossRef]
39. Peng X, Wei Z, Liu S, Liu L, Jiang H, Yu L, et al. Effect and mechanism of fisetin against the development of metabolic dysfunction-associated fatty liver disease. Sci Rep. 2025;15(1):35766. doi:10.1038/s41598-025-96848-0. [Google Scholar] [PubMed] [CrossRef]
40. Shen TK, Vignane T, Gilglioni EH, Traini L, Kalaitsidou E, Conan P, et al. Metabolic dysfunction-associated steatohepatitis reduces hepatic H(2)S-producing enzymes altering persulfidome composition. Redox Biol. 2025;86:103809. doi:10.1016/j.redox.2025.103809. [Google Scholar] [PubMed] [CrossRef]
41. Vaziri ND, Dicus M, Ho ND, Boroujerdi-Rad L, Sindhu RK. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int. 2003;63(1):179–85. doi:10.1046/j.1523-1755.2003.00702.x. [Google Scholar] [PubMed] [CrossRef]
42. Karamouzis I, Sarafidis PA, Karamouzis M, Iliadis S, Haidich AB, Sioulis A, et al. Increase in oxidative stress but not in antioxidant capacity with advancing stages of chronic kidney disease. Am J Nephrol. 2008;28(3):397–404. doi:10.1159/000112413. [Google Scholar] [PubMed] [CrossRef]
43. Petrosillo G, Portincasa P, Grattagliano I, Casanova G, Matera M, Ruggiero FM, et al. Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim Biophys Acta. 2007;1767(10):1260–7. doi:10.1016/j.bbabio.2007.07.011. [Google Scholar] [PubMed] [CrossRef]
44. Aparicio-Trejo OE, Rojas-Morales P, Avila-Rojas SH, León-Contreras JC, Hernández-Pando R, Jiménez-Uribe AP, et al. Temporal alterations in mitochondrial β-oxidation and oxidative stress aggravate chronic kidney disease development in 5/6 nephrectomy induced renal damage. Int J Mol Sci. 2020;21(18):6512. doi:10.3390/ijms21186512. [Google Scholar] [PubMed] [CrossRef]
45. Kühlbrandt W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015;13:89. doi:10.1186/s12915-015-0201-x. [Google Scholar] [PubMed] [CrossRef]
46. Zhao Y, Zhou Y, Wang D, Huang Z, Xiao X, Zheng Q, et al. Mitochondrial dysfunction in metabolic dysfunction fatty liver disease (MAFLD). Int J Mol Sci. 2023;24(24):17514. doi:10.3390/ijms242417514. [Google Scholar] [PubMed] [CrossRef]
47. Li Y, Wu J, Yang M, Wei L, Wu H, Wang Q, et al. Physiological evidence of mitochondrial permeability transition pore opening caused by lipid deposition leading to hepatic steatosis in db/db mice. Free Radic Biol Med. 2021;162:523–32. doi:10.1016/j.freeradbiomed.2020.11.009. [Google Scholar] [PubMed] [CrossRef]
48. Lindblom RSJ, Higgins GC, Nguyen TV, Arnstein M, Henstridge DC, Granata C, et al. Delineating a role for the mitochondrial permeability transition pore in diabetic kidney disease by targeting cyclophilin D. Clin Sci. 2020;134(2):239–59. doi:10.1042/CS20190787. [Google Scholar] [PubMed] [CrossRef]
49. Nesci S, Rubattu S. UCP2, a member of the mitochondrial uncoupling proteins: an overview from physiological to pathological roles. Biomedicines. 2024;12(6):1307. doi:10.3390/biomedicines12061307. [Google Scholar] [PubMed] [CrossRef]
50. Papu John AS, Kundu S, Pushpakumar S, Amin M, Tyagi SC, Sen U. Hydrogen sulfide inhibits Ca2+-induced mitochondrial permeability transition pore opening in type-1 diabetes. Am J Physiol Endocrinol Metab. 2019;317(2):E269–83. doi:10.1152/ajpendo.00251.2018. [Google Scholar] [PubMed] [CrossRef]
51. Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. 2014;15(10):634–46. doi:10.1038/nrm3877. [Google Scholar] [PubMed] [CrossRef]
52. Galvan DL, Green NH, Danesh FR. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017;92(5):1051–7. doi:10.1016/j.kint.2017.05.034. [Google Scholar] [PubMed] [CrossRef]
53. Jang S, Javadov S. OPA1 regulates respiratory super complexes assembly: the role of mitochondrial swelling. Mitochondrion. 2020;51:30–9. doi:10.1016/j.mito.2019.11.006. [Google Scholar] [PubMed] [CrossRef]
54. Vezza T, Díaz-Pozo P, Canet F, de Marañón AM, Abad-Jiménez Z, García-Gargallo C, et al. The role of mitochondrial dynamic dysfunction in age-associated type 2 diabetes. World J Mens Health. 2022;40(3):399–411. doi:10.5534/wjmh.210146. [Google Scholar] [PubMed] [CrossRef]
55. Zheng P, Ma W, Gu Y, Wu H, Bian Z, Liu N, et al. High-fat diet causes mitochondrial damage and downregulation of mitofusin-2 and optic atrophy-1 in multiple organs. J Clin Biochem Nutr. 2023;73(1):61–76. doi:10.3164/jcbn.22-73. [Google Scholar] [PubMed] [CrossRef]
56. Yoo J, Cheol Hwang Y. 1599-P: regulation of mitochondrial dynamics ameliorates hepatic steatosis through TFEB activation. Diabetes. 2023;72(Suppl 1):1599–P. doi:10.2337/db23-1599-p. [Google Scholar] [CrossRef]
57. Li L, Martin-Levilain J, Jiménez-Sánchez C, Karaca M, Foti M, Martinou JC, et al. In vivo stabilization of OPA1 in hepatocytes potentiates mitochondrial respiration and gluconeogenesis in a prohibitin-dependent way. J Biol Chem. 2019;294(34):12581–98. doi:10.1074/jbc.RA119.007601. [Google Scholar] [PubMed] [CrossRef]
58. Lee K, Haddad A, Osme A, Kim C, Borzou A, Ilchenko S, et al. Hepatic mitochondrial defects in a nonalcoholic fatty liver disease mouse model are associated with increased degradation of oxidative phosphorylation subunits. Mol Cell Proteomics. 2018;17(12):2371–86. doi:10.1074/mcp.RA118.000961. [Google Scholar] [PubMed] [CrossRef]
59. Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014;19(4):630–41. doi:10.1016/j.cmet.2014.03.011. [Google Scholar] [PubMed] [CrossRef]
60. Sookoian S, Flichman D, Scian R, Rohr C, Dopazo H, Gianotti TF, et al. Mitochondrial genome architecture in non-alcoholic fatty liver disease. J Pathol. 2016;240(4):437–49. doi:10.1002/path.4803. [Google Scholar] [PubMed] [CrossRef]
61. Galloway CA, Lee H, Nejjar S, Jhun BS, Yu T, Hsu W, et al. Transgenic control of mitochondrial fission induces mitochondrial uncoupling and relieves diabetic oxidative stress. Diabetes. 2012;61(8):2093–104. doi:10.2337/db11-1640. [Google Scholar] [PubMed] [CrossRef]
62. Zhan M, Usman IM, Sun L, Kanwar YS. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J Am Soc Nephrol. 2015;26(6):1304–21. doi:10.1681/ASN.2014050457. [Google Scholar] [PubMed] [CrossRef]
63. Prieto-Carrasco R, García-Arroyo FE, Aparicio-Trejo OE, Rojas-Morales P, León-Contreras JC, Hernández-Pando R, et al. Progressive reduction in mitochondrial mass is triggered by alterations in mitochondrial biogenesis and dynamics in chronic kidney disease induced by 5/6 nephrectomy. Biology. 2021;10(5):349. doi:10.3390/biology10050349. [Google Scholar] [PubMed] [CrossRef]
64. Song Z, Xia Y, Shi L, Zha H, Huang J, Xiang X, et al. Inhibition of Drp1-Fis1 interaction alleviates aberrant mitochondrial fragmentation and acute kidney injury. Cell Mol Biol Lett. 2024;29(1):31. doi:10.1186/s11658-024-00553-1. [Google Scholar] [PubMed] [CrossRef]
65. Ding XQ, Jian TY, Gai YN, Niu GT, Liu Y, Meng XH, et al. Chicoric acid attenuated renal tubular injury in HFD-induced chronic kidney disease mice through the promotion of mitophagy via the Nrf2/PINK/parkin pathway. J Agric Food Chem. 2022;70(9):2923–35. doi:10.1021/acs.jafc.1c07795. [Google Scholar] [PubMed] [CrossRef]
66. Coughlan MT, Nguyen TV, Penfold SA, Higgins GC, Thallas-Bonke V, Tan SM, et al. Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin Sci. 2016;130(9):711–20. doi:10.1042/CS20150838. [Google Scholar] [PubMed] [CrossRef]
67. Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393(7):547–64. doi:10.1515/hsz-2012-0119. [Google Scholar] [PubMed] [CrossRef]
68. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. doi:10.1038/nrm3028. [Google Scholar] [PubMed] [CrossRef]
69. Tang C, He L, Liu J, Dong Z. Mitophagy: basic mechanism and potential role in kidney diseases. Kidney Dis. 2015;1(1):71–9. doi:10.1159/000381510. [Google Scholar] [PubMed] [CrossRef]
70. Smiles WJ, Ovens AJ, Kemp BE, Galic S, Petersen J, Oakhill JS. New developments in AMPK and mTORC1 cross-talk. Essays Biochem. 2024;68(3):321–36. doi:10.1042/ebc20240007. [Google Scholar] [PubMed] [CrossRef]
71. Shin S, Kim J, Lee JY, Kim J, Oh CM. Mitochondrial quality control: its role in metabolic dysfunction-associated steatotic liver disease (MASLD). J Obes Metab Syndr. 2023;32(4):289–302. doi:10.7570/jomes23054. [Google Scholar] [PubMed] [CrossRef]
72. Li W, Cai Z, Schindler F, Afjehi-Sadat L, Montsch B, Heffeter P, et al. Elevated PINK1/parkin-dependent mitophagy and boosted mitochondrial function mediate protection of HepG2 cells from excess palmitic acid by hesperetin. J Agric Food Chem. 2024;72(23):13039–53. doi:10.1021/acs.jafc.3c09132. [Google Scholar] [PubMed] [CrossRef]
73. Liu P, Lin H, Xu Y, Zhou F, Wang J, Liu J, et al. Frataxin-mediated PINK1-parkin-dependent mitophagy in hepatic steatosis: the protective effects of quercetin. Mol Nutr Food Res. 2018;62(16):e1800164. doi:10.1002/mnfr.201800164. [Google Scholar] [PubMed] [CrossRef]
74. Huang Z, Luo L, Xiao Z, Xiong M, Wen Z. Omentin-1 mitigates non-alcoholic fatty liver disease by preserving autophagy through AMPKα/mTOR signaling pathway. Sci Rep. 2024;14(1):31464. doi:10.1038/s41598-024-83112-0. [Google Scholar] [PubMed] [CrossRef]
75. Zhou T, Chang L, Luo Y, Zhou Y, Zhang J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. 2019;21:101120. doi:10.1016/j.redox.2019.101120. [Google Scholar] [PubMed] [CrossRef]
76. Wang Y, Tang C, Cai J, Chen G, Zhang D, Zhang Z, et al. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018;9(11):1113. doi:10.1038/s41419-018-1152-2. [Google Scholar] [PubMed] [CrossRef]
77. Bellezza I, Giambanco I, Minelli A, Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta BBA Mol Cell Res. 2018;1865(5):721–33. doi:10.1016/j.bbamcr.2018.02.010. [Google Scholar] [PubMed] [CrossRef]
78. Das F, Bera A, Ghosh-Choudhury N, Sataranatarajan K, Kamat A, Kasinath BS, et al. High glucose-stimulated enhancer of zeste homolog-2 (EZH2) forces suppression of deptor to cause glomerular mesangial cell pathology. Cell Signal. 2021;86:110072. doi:10.1016/j.cellsig.2021.110072. [Google Scholar] [PubMed] [CrossRef]
79. Abergel E, Pri-Chen H, Wallach-Dayan S, Hinden L, Tam J, Volovelsky O, et al. Targeting the endocannabinoid system to suppress mTORC1 hyperactivation in TSC-associated kidney disease. Am J Physiol Renal Physiol. 2025;329(3):F325–34. doi:10.1152/ajprenal.00097.2025. [Google Scholar] [PubMed] [CrossRef]
80. Zhou J, Sun DQ, Targher G, Byrne DC, Lee BW, Hamaguchi M, et al. Metabolic dysfunction-associated fatty liver disease increases risk of chronic kidney disease: a systematic review and meta-analysis. eGastroenterology. 2023;1(1):e100005. doi:10.1136/egastro-2023-100005. [Google Scholar] [PubMed] [CrossRef]
81. Deprince A, Haas JT, Staels B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol Metab. 2020;42:101092. doi:10.1016/j.molmet.2020.101092. [Google Scholar] [PubMed] [CrossRef]
82. Nishi H, Higashihara T, Inagi R. Lipotoxicity in kidney, heart, and skeletal muscle dysfunction. Nutrients. 2019;11(7):1664. doi:10.3390/nu11071664. [Google Scholar] [PubMed] [CrossRef]
83. Chen Y, Yan Q, Lv M, Song K, Dai Y, Huang Y, et al. Involvement of FATP2-mediated tubular lipid metabolic reprogramming in renal fibrogenesis. Cell Death Dis. 2020;11(11):994. doi:10.1038/s41419-020-03199-x. [Google Scholar] [PubMed] [CrossRef]
84. Khan S, Gaivin R, Abramovich C, Boylan M, Calles J, Schelling JR. Fatty acid transport protein-2 regulates glycemic control and diabetic kidney disease progression. JCI Insight. 2020;5(15):e136845. doi:10.1172/jci.insight.136845. [Google Scholar] [PubMed] [CrossRef]
85. Han Y, Su Y, Han M, Liu Y, Shi Q, Li X, et al. Ginsenoside Rg1 attenuates glomerular fibrosis by inhibiting CD36/TRPC6/NFAT2 signaling in type 2 diabetes mellitus mice. J Ethnopharmacol. 2023;302(Pt A):115923. doi:10.1016/j.jep.2022.115923. [Google Scholar] [PubMed] [CrossRef]
86. Xu M, Zhou H, Hu P, Pan Y, Wang S, Liu L, et al. Identification and validation of immune and oxidative stress-related diagnostic markers for diabetic nephropathy by WGCNA and machine learning. Front Immunol. 2023;14:1084531. doi:10.3389/fimmu.2023.1084531. [Google Scholar] [PubMed] [CrossRef]
87. Yen YH, Chen JB, Cheng BC, Chen JF, Chang KC, Tseng PL, et al. Using controlled attenuation parameter combined with ultrasound to survey non-alcoholic fatty liver disease in hemodialysis patients: a prospective cohort study. PLoS One. 2017;12(4):e0176027. doi:10.1371/journal.pone.0176027. [Google Scholar] [PubMed] [CrossRef]
88. Stoica RA, Tribus LC, Marin RI, David T, Preda CM, Bica IC, et al. Non-alcoholic fatty liver disease in diabetes mellitus patients on chronic hemodialysis—a case series addressing cardiovascular and mortality risks. Front Clin Diabetes Healthc. 2023;4:1113666. doi:10.3389/fcdhc.2023.1113666. [Google Scholar] [PubMed] [CrossRef]
89. Lisowska-Myjak B. Uremic toxins and their effects on multiple organ systems. Nephron Clin Pract. 2014;128(3–4):303–11. doi:10.1159/000369817. [Google Scholar] [PubMed] [CrossRef]
90. Raillon LA, Florens N, Payelle F, Martin M, Soulère L, Yi D, et al. Medium chain fatty acids are potent binding competitors to improve protein-bound uremic toxin clearance during hemodialysis. Kidney Int. 2025;108(3):411–26. doi:10.1016/j.kint.2025.06.004. [Google Scholar] [PubMed] [CrossRef]
91. Santana Machado T, Poitevin S, Paul P, McKay N, Jourde-Chiche N, Legris T, et al. Indoxyl sulfate upregulates liver P-glycoprotein expression and activity through aryl hydrocarbon receptor signaling. J Am Soc Nephrol. 2018;29(3):906–18. doi:10.1681/ASN.2017030361. [Google Scholar] [PubMed] [CrossRef]
92. Bush KT, Singh P, Nigam SK. Gut-derived uremic toxin handling in vivo requires OAT-mediated tubular secretion in chronic kidney disease. JCI Insight. 2020;5(7):e133817. doi:10.1172/jci.insight.133817. [Google Scholar] [PubMed] [CrossRef]
93. Yabuuchi N, Hou H, Nao G, Narita Y, Jono H, Saito H. Suppressed hepatic production of indoxyl sulfate attenuates cisplatin-induced acute kidney injury in sulfotransferase 1a1-deficient mice. Int J Mol Sci. 2021;22(4):1764. doi:10.3390/ijms22041764. [Google Scholar] [PubMed] [CrossRef]
94. Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol. 2023;23(3):159–73. doi:10.1038/s41577-022-00760-x. [Google Scholar] [PubMed] [CrossRef]
95. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–14. doi:10.1016/j.immuni.2012.01.009. [Google Scholar] [PubMed] [CrossRef]
96. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–7. doi:10.1038/nature14156. [Google Scholar] [PubMed] [CrossRef]
97. Kent AC, El Baradie KBY, Hamrick MW. Targeting the mitochondrial permeability transition pore to prevent age-associated cell damage and neurodegeneration. Oxid Med Cell Longev. 2021;2021:6626484. doi:10.1155/2021/6626484. [Google Scholar] [PubMed] [CrossRef]
98. Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011;208(3):417–20. doi:10.1084/jem.20110367. [Google Scholar] [PubMed] [CrossRef]
99. Yang M, Geng CA, Liu X, Guan M. Lipid disorders in NAFLD and chronic kidney disease. Biomedicines. 2021;9(10):1405. doi:10.3390/biomedicines9101405. [Google Scholar] [PubMed] [CrossRef]
100. Bogdan RG, Boicean A, Anderco P, Ichim C, Iliescu-Glaja M, Todor SB, et al. From liver to kidney: the overlooked burden of nonalcoholic fatty liver disease in chronic kidney disease. J Clin Med. 2025;14(7):2486. doi:10.3390/jcm14072486. [Google Scholar] [PubMed] [CrossRef]
101. Fularski P, Czarnik W, Frankenstein H, Gąsior M, Młynarska E, Rysz J, et al. Unveiling selected influences on chronic kidney disease development and progression. Cells. 2024;13(9):751. doi:10.3390/cells13090751. [Google Scholar] [PubMed] [CrossRef]
102. Alvarado-Ojeda ZA, Trejo-Moreno C, Ferat-Osorio E, Méndez-Martínez M, Fragoso G, Rosas-Salgado G. Role of angiotensin II in non-alcoholic steatosis development. Arch Med Res. 2024;55(3):102986. doi:10.1016/j.arcmed.2024.102986. [Google Scholar] [PubMed] [CrossRef]
103. Rad NK, Heydari Z, Tamimi AH, Zahmatkesh E, Shpichka A, Barekat M, et al. Review on kidney-liver crosstalk: pathophysiology of their disorders. Cell J. 2024;26(2):98–111. [Google Scholar] [PubMed]
104. Barbosa GSB, Câmara NOS, Ledesma FL, Duarte Neto AN, Dias CB. Vascular injury in glomerulopathies: the role of the endothelium. Front Nephrol. 2024;4:1396588. doi:10.3389/fneph.2024.1396588. [Google Scholar] [PubMed] [CrossRef]
105. Bataller R, Gäbele E, Schoonhoven R, Morris T, Lehnert M, Yang L, et al. Prolonged in fusi on of angiotensin II into normal rats induces stellate cell activation and proinflammatory events in liver. Am J Physiol Gastrointest Liver Physiol. 2003;285(3):G642–51. doi:10.1152/ajpgi.00037.2003. [Google Scholar] [PubMed] [CrossRef]
106. Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Investig. 2003;112(9):1383–94. doi:10.1172/JCI18212. [Google Scholar] [PubMed] [CrossRef]
107. Fukai T, Ushio-Fukai M. Cross-talk between NADPH oxidase and mitochondria: role in ROS signaling and angiogenesis. Cells. 2020;9(8):1849. doi:10.3390/cells9081849. [Google Scholar] [PubMed] [CrossRef]
108. Dikalov SI, Nazarewicz RR. Angiotensin II-induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal. 2013;19(10):1085–94. doi:10.1089/ars.2012.4604. [Google Scholar] [PubMed] [CrossRef]
109. Poussin C, Ibberson M, Hall D, Ding J, Soto J, Abel ED, et al. Oxidative phosphorylation flexibility in the liver of mice resistant to high-fat diet-induced hepatic steatosis. Diabetes. 2011;60(9):2216–24. doi:10.2337/db11-0338. [Google Scholar] [PubMed] [CrossRef]
110. Castañé H, Jiménez-Franco A, Hernández-Aguilera A, Martínez-Navidad C, Cambra-Cortés V, Onoiu AI, et al. Multi-omics profiling reveals altered mitochondrial metabolism in adipose tissue from patients with metabolic dysfunction-associated steatohepatitis. EBioMedicine. 2025;111:105532. doi:10.1016/j.ebiom.2024.105532. [Google Scholar] [PubMed] [CrossRef]
111. Ruggiero C, Ehrenshaft M, Cleland E, Stadler K. High-fat diet induces an initial adaptation of mitochondrial bioenergetics in the kidney despite evident oxidative stress and mitochondrial ROS production. Am J Physiol Endocrinol Metab. 2011;300(6):E1047–58. doi:10.1152/ajpendo.00666.2010. [Google Scholar] [PubMed] [CrossRef]
112. Prem PN, Kurian GA. High-fat diet increased oxidative stress and mitochondrial dysfunction induced by renal ischemia-reperfusion injury in rat. Front Physiol. 2021;12:715693. doi:10.3389/fphys.2021.715693. [Google Scholar] [PubMed] [CrossRef]
113. Chen S, Chen J, Li S, Guo F, Li A, Wu H, et al. High-fat diet-induced renal proximal tubular inflammatory injury: emerging risk factor of chronic kidney disease. Front Physiol. 2021;12:786599. doi:10.3389/fphys.2021.786599. [Google Scholar] [PubMed] [CrossRef]
114. He WJ, Li C, Huang Z, Geng S, Rao VS, Kelly TN, et al. Association of mitochondrial DNA copy number with risk of progression of kidney disease. Clin J Am Soc Nephrol. 2022;17(7):966–75. doi:10.2215/cjn.15551121. [Google Scholar] [PubMed] [CrossRef]
115. Ma X, Wei X, Niu M, Zhang C, Peng Z, Liu W, et al. Disruption of mitochondrial dynamics and stasis leads to liver injury and tumorigenesis [Internet]. 2025 [cited 2025 Nov 13]. Available from: https://www.biorxiv.org/content/10.1101/2025.02.11.637688v1. [Google Scholar]
116. Yuan L, Yuan Y, Liu F, Li L, Liu J, Chen Y, et al. PGC-1α alleviates mitochondrial dysfunction via TFEB-mediated autophagy in cisplatin-induced acute kidney injury. Aging. 2021;13(6):8421–39. doi:10.18632/aging.202653. [Google Scholar] [PubMed] [CrossRef]
117. Spoto B, Politi C, Pizzini P, Parlongo RM, Testa A, Mobrici M, et al. 8-hydroxy-2′-deoxyguanosine, a biomarker of oxidative DNA injury, in diabetic kidney disease. Nutr Metab Cardiovasc Dis. 2025;35(2):103722. doi:10.1016/j.numecd.2024.08.015. [Google Scholar] [PubMed] [CrossRef]
118. Rana R, Huirem RS, Kant R, Chauhan K, Sharma S, Yashavarddhan MH, et al. Cytochrome C as a potential clinical marker for diagnosis and treatment of glioma. Front Oncol. 2022;12:960787. doi:10.3389/fonc.2022.960787. [Google Scholar] [PubMed] [CrossRef]
119. Wang T, Sun F, Li C, Nan P, Song Y, Wan X, et al. MTA1, a novel ATP synthase complex modulator, enhances colon cancer liver metastasis by driving mitochondrial metabolism reprogramming. Adv Sci. 2023;10(25):2300756. doi:10.1002/advs.202300756. [Google Scholar] [PubMed] [CrossRef]
120. Punetha M, Saini S, Chaudhary S, Bala R, Sharma M, Kumar P, et al. Mitochondria-targeted antioxidant MitoQ ameliorates ROS production and improves cell viability in cryopreserved buffalo fibroblasts. Tissue Cell. 2023;82:102067. doi:10.1016/j.tice.2023.102067. [Google Scholar] [PubMed] [CrossRef]
121. Yang J, Suo H, Song J. Protective role of mitoquinone against impaired mitochondrial homeostasis in metabolic syndrome. Crit Rev Food Sci Nutr. 2021;61(22):3857–75. doi:10.1080/10408398.2020.1809344. [Google Scholar] [PubMed] [CrossRef]
122. Smith RAJ, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci. 2010;1201(1):96–103. doi:10.1111/j.1749-6632.2010.05627.x. [Google Scholar] [PubMed] [CrossRef]
123. Fouret G, Tolika E, Lecomte J, Bonafos B, Aoun M, Murphy MP, et al. The mitochondrial-targeted antioxidant, MitoQ, increases liver mitochondrial cardiolipin content in obesogenic diet-fed rats. Biochim Biophys Acta. 2015;1847(10):1025–35. doi:10.1016/j.bbabio.2015.05.019. [Google Scholar] [PubMed] [CrossRef]
124. Kirkman DL, Stock JM, Shenouda N, Bohmke NJ, Kim Y, Kidd J, et al. Effects of a mitochondrial-targeted ubiquinol on vascular function and exercise capacity in chronic kidney disease: a randomized controlled pilot study. Am J Physiol Renal Physiol. 2023;325(4):F448–56. doi:10.1152/ajprenal.00067.2023. [Google Scholar] [PubMed] [CrossRef]
125. Pharaoh G, Kamat V, Kannan S, Stuppard RS, Whitson J, Martin-Perez M, et al. Elamipretide improves ADP sensitivity in aged mitochondria by increasing uptake through the adenine nucleotide translocator (ANT) [Internet]. 2023 [cited 2025 Nov 13]. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC9915686/. [Google Scholar]
126. Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. 2013;24(8):1250–61. doi:10.1681/ASN.2012121216. [Google Scholar] [PubMed] [CrossRef]
127. Tung C, Varzideh F, Farroni E, Mone P, Kansakar U, Jankauskas SS, et al. Elamipretide: a review of its structure, mechanism of action, and therapeutic potential. Int J Mol Sci. 2025;26(3):944. doi:10.3390/ijms26030944. [Google Scholar] [PubMed] [CrossRef]
128. Bhatti JS, Tamarai K, Kandimalla R, Manczak M, Yin X, Ramasubramanian B, et al. Protective effects of a mitochondria-targeted small peptide SS31 against hyperglycemia-induced mitochondrial abnormalities in the liver tissues of diabetic mice, Tallyho/JngJ mice. Mitochondrion. 2021;58(Suppl 1):49–58. doi:10.1016/j.mito.2021.02.007. [Google Scholar] [PubMed] [CrossRef]
129. Patel P, Mendoza A, Ramirez D, Robichaux D, Molkentin JD, Karch J. The adenine nucleotide translocase family underlies cardiac ischemia-reperfusion injury through the mitochondrial permeability pore independently of cyclophilin D. Sci Adv. 2024;10(50):eadp7444. doi:10.1126/sciadv.adp7444. [Google Scholar] [PubMed] [CrossRef]
130. Guan N, Ren YL, Liu XY, Zhang Y, Pei P, Zhu SN, et al. Protective role of cyclosporine A and minocycline on mitochondrial disequilibrium-related podocyte injury and proteinuria occurrence induced by adriamycin. Nephrol Dial Transplant. 2015;30(6):957–69. doi:10.1093/ndt/gfv015. [Google Scholar] [PubMed] [CrossRef]
131. Dong Y, Wu X, Li J, Li J, Lu Y, He C, et al. Phloretin suppresses the mPTP abnormal opening via the SHP-2/JAK2/BAX axis to ameliorate nonalcoholic steatohepatitis. J Agric Food Chem. 2025;73(42):26700–13. doi:10.1021/acs.jafc.5c05787. [Google Scholar] [PubMed] [CrossRef]
132. Adhikari A, Mondal S, Chatterjee T, Das M, Biswas P, Ghosh R, et al. Redox nanomedicine ameliorates chronic kidney disease (CKD) by mitochondrial reconditioning in mice. Commun Biol. 2021;4(1):1013. doi:10.1038/s42003-021-02546-8. [Google Scholar] [PubMed] [CrossRef]
133. de Luxán-Delgado B, Potes Y, Rubio-González A, Solano JJ, Boga JA, Antuña E, et al. Melatonin alleviates liver mitochondrial dysfunction in leptin-deficient mice. Int J Mol Sci. 2024;25(16):8677. doi:10.3390/ijms25168677. [Google Scholar] [PubMed] [CrossRef]
134. Bahrami M, Cheraghpour M, Jafarirad S, Alavinejad P, Asadi F, Hekmatdoost A, et al. The effect of melatonin on treatment of patients with non-alcoholic fatty liver disease: a randomized double blind clinical trial. Complement Ther Med. 2020;52(2):102452. doi:10.1016/j.ctim.2020.102452. [Google Scholar] [PubMed] [CrossRef]
135. Agil A, Chayah M, Visiedo L, Navarro-Alarcon M, Rodríguez Ferrer JM, Tassi M, et al. Melatonin improves mitochondrial dynamics and function in the kidney of zücker diabetic fatty rats. J Clin Med. 2020;9(9):2916. doi:10.3390/jcm9092916. [Google Scholar] [PubMed] [CrossRef]
136. Yang C, Xiao C, Ding Z, Zhai X, Liu J, Yu M. Canagliflozin mitigates diabetic cardiomyopathy through enhanced PINK1-parkin mitophagy. Int J Mol Sci. 2024;25(13):7008. doi:10.3390/ijms25137008. [Google Scholar] [PubMed] [CrossRef]
137. Heerspink HJL, Stefánsson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, et al. Dapagliflozin in patients with chronic kidney disease. N Engl J Med. 2020;383(15):1436–46. doi:10.1056/nejmoa2024816. [Google Scholar] [PubMed] [CrossRef]
138. Kuchay MS, Krishan S, Mishra SK, Farooqui KJ, Singh MK, Wasir JS, et al. Effect of empagliflozin on liver fat in patients with type 2 diabetes and nonalcoholic fatty liver disease: a randomized controlled trial (E-LIFT trial). Diabetes Care. 2018;41(8):1801–8. doi:10.2337/dc18-0165. [Google Scholar] [PubMed] [CrossRef]
139. Liu Y, Zhang M, Wang Y. Induction of autophagy as a therapeutic breakthrough for NAFLD: current evidence and perspectives. Biology. 2025;14(8):989. doi:10.3390/biology14080989. [Google Scholar] [PubMed] [CrossRef]
140. Ruan L, Chen J, Du C, Lu H, Zhang J, Cai X, et al. Mitochondrial temperature-responsive drug delivery reverses drug resistance in lung cancer. Bioact Mater. 2021;13:191–9. doi:10.1016/j.bioactmat.2021.10.045. [Google Scholar] [PubMed] [CrossRef]
141. Yu H, Jin F, Liu D, Shu G, Wang X, Qi J, et al. ROS-responsive nano-drug delivery system combining mitochondria-targeting ceria nanoparticles with atorvastatin for acute kidney injury. Theranostics. 2020;10(5):2342–57. doi:10.7150/thno.40395. [Google Scholar] [PubMed] [CrossRef]
142. Casals G, Perramón M, Casals E, Portolés I, Fernández-Varo G, Morales-Ruiz M, et al. Cerium oxide nanoparticles: a new therapeutic tool in liver diseases. Antioxidants. 2021;10(5):660. doi:10.3390/antiox10050660. [Google Scholar] [PubMed] [CrossRef]
143. Stefano JT, Duarte SMB, Ribeiro Leite Altikes RG, Oliveira CP. Non-pharmacological management options for MAFLD: a practical guide. Ther Adv Endocrinol Metab. 2023;14:20420188231160394. doi:10.1177/20420188231160394. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools