
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Exploring the Latest Developments in Natural Killer (NK) Cell-Based Therapies for Diffuse Intrinsic Pontine Glioma (DIPG)
ImmuneLink, LLC, Riverside, CA 92508, USA
* Corresponding Author: Kawaljit Kaur. Email:
(This article belongs to the Special Issue: Novel Targeted Therapy in Oncology)
BIOCELL 2026, 50(3), 4 https://doi.org/10.32604/biocell.2025.073340
Received 16 September 2025; Accepted 20 November 2025; Issue published 23 March 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Diffuse intrinsic pontine glioma (DIPG) is a pediatric brainstem tumor with a very poor prognosis, characterized by immunosuppressive tumor microenvironment (TME) that limits immune infiltration, including a significant reduction in circulating natural killer (NK) cells. This drop in NK cell levels and activity may promote tumor growth and immune evasion, making NK cells a promising target for immunotherapy. NK cells can attack and eliminate DIPG tumor cells, including glioma stem cells, while counteracting certain immune evasion strategies. Although the DIPG microenvironment and blood-brain barrier present challenges, NK cell-based therapies have shown encouraging tumor control and survival benefits in animal models with promising safety results. Current clinical trials for DIPG mostly focus on chimeric antigen receptor (CAR)-T cells targeting disialoganglioside (GD2) and cluster of differentiation 276 (CD276 or B7-H3) antigens with early signs of success, while NK cell therapies, such as CAR-NK cells, are still in preclinical or early stages, requiring further development. The tumor’s immunosuppressive nature poses challenges that may need combination strategies or immune priming. Despite these obstacles, NK cell-based immunotherapy is an exciting and growing field. Upcoming clinical trials emphasize the potential for NK cell therapies to play a critical role in treating this aggressive pediatric brain cancer.Keywords
1 Introduction and Background: Diffuse Intrinsic Pontine Glioma
Diffuse intrinsic pontine glioma (DIPG) is an aggressive pediatric brain tumor that develops in the pons, a part of the brainstem responsible for essential functions like breathing, heart rate, and blood pressure, as well as controlling vision, hearing, and movement [1–4]. DIPG originates from monopotent stem cells derived from oligodendrocyte precursor cells, which can self-renew and produce myelinating oligodendrocytes [5]. The glioma growth factor neuroligin-3 (NLGN3) in the DIPG tumor microenvironment significantly promotes tumor growth [1,2]. These tumors often impact deep midline brain structures in young children, likely tied to epigenetic regulation in early brain development [3]. Over 85% of DIPG tumors involve a lysine-to-methionine mutation at position 27 in the histone (H3K27M), which drives cancer growth [3–5]. The H3K27M mutation in histone H3 disrupts the polycomb repressive complex 2 (PRC2), hindering proper trimethylation at lysine 27 (H3K27me3) [6–8]. This causes a global decrease in transcriptional repression marks and an increase in activating acetylation marks (H3K27ac), enhancing chromatin accessibility and triggering oncogenic pathways [6,9,10]. Tumors with this mutation are classified as grade 4 under the World Health Organization’s H3 K27-altered diffuse midline glioma category [8]. DIPG’s challenging location and interaction with the blood-brain barrier (BBB) make it particularly severe, with 150–400 new cases diagnosed annually in the United States [11,12]. Tragically, only 10% of children diagnosed survive beyond two years, with most cases occurring in children under age 9 [3,13–15]. Symptoms, which can appear suddenly, include vision issues, difficulty talking or swallowing, nausea, weakness, balance problems, trouble walking, and behavioral changes [16]. Several treatments for DIPG have been explored, but success rates remain minimal [16]. Due to its delicate location, treatment options like surgery, chemotherapy, and radiation are limited, underscoring the urgent need for advancements in immune cell-based therapies to address the tumor’s immunosuppressive microenvironment and poor prognosis [3,4,12,17,18].
Research indicates that natural killer (NK) cells, derived from the peripheral blood of healthy individuals, can effectively target and differentiate glioma tumors [19–21]. These cytotoxic lymphocytes, which make up 5%–15% of the immune cells in healthy people, serve as a crucial first line of defense against tumor growth. NK cells hold therapeutic potential due to their innate ability to attack cells lacking or mutating major histocompatibility complex (MHC) class I [22]. They recognize and kill tumor cells without prior activation, relying on a balance of stimulatory and inhibitory receptors. NK cells play a significant role in cancer inhibition through direct cytotoxicity, antibody-dependent cellular cytotoxicity (ADCC), and by regulating other immune effectors with their cytokines and chemokines [23–25]. They act swiftly to eliminate threats, both directly and by modulating immune responses. Preclinical studies have shown that NK cells are effective against brain tumors, with glioblastoma multiforme (GBM) responding well to IL-2 or IL-15-activated NK cells [26,27]. NK cells combined with monoclonal antibodies have also shown success in treating GBM [28]. However, cancer patients’ NK cells often face challenges like tumor-induced inactivation and short lifespans, which reduce their therapeutic viability without enhancements [29,30].
Though advancements in therapeutics have been made, DIPG remains difficult to treat due to its location, genetic complexity, and the intact BBB [31,32]. Current clinical trials focus on safety, pharmacokinetics, and efficacy, though further evaluation is required. While a definitive cure is lacking, recent trials integrating novel delivery methods, targeted therapies, and immunotherapy show promise. Effective drug delivery techniques like convection-enhanced delivery (CED), intra-arterial infusion, and MR-guided focused ultrasound (MRgFUS) are essential for success [31,33]. Analyses of the tumor microenvironment reveal that DIPG tumors are immunologically “cold”, with little infiltration of NK cells and other lymphocytes [34]. Clinical studies show that DIPG patients have much lower peripheral NK cell levels and functions compared to healthy controls [21,35]. Decreased number and activity of NK cells may play a role in DIPG tumor development and immune evasion [35]. To overcome these issues, allogeneic NK cell therapies are being explored in clinical trials as standalone or combination treatments [36–39].
This review focuses on the challenges of DIPG and the obstacles they pose for effective treatments like immunotherapy. NK cell-based therapies, which utilize innate immunity and low toxicity, offer a promising and innovative approach, especially if issues like tumor infiltration and persistence are resolved. Several clinical trials are investigating NK-cell-based CAR constructs and antibody combinations targeting DIPG antigens. Moreover, advancements in crossing the BBB and altering the tumor microenvironment are vital for better outcomes. Progress in NK cell engineering, delivery systems, and combined immunotherapy strategies brings hope for future breakthroughs in DIPG treatment.
The relationship between DIPG and its tumor microenvironment (TME) presents challenges and opportunities for treatment. Key targets are genetic alterations, neurotransmitters, immune checkpoints, and ligand-receptor interactions. Identifying and targeting specific molecular pathways and immune interactions within the TME could lead to improved therapies for this aggressive pediatric brain tumor (Fig. 1).
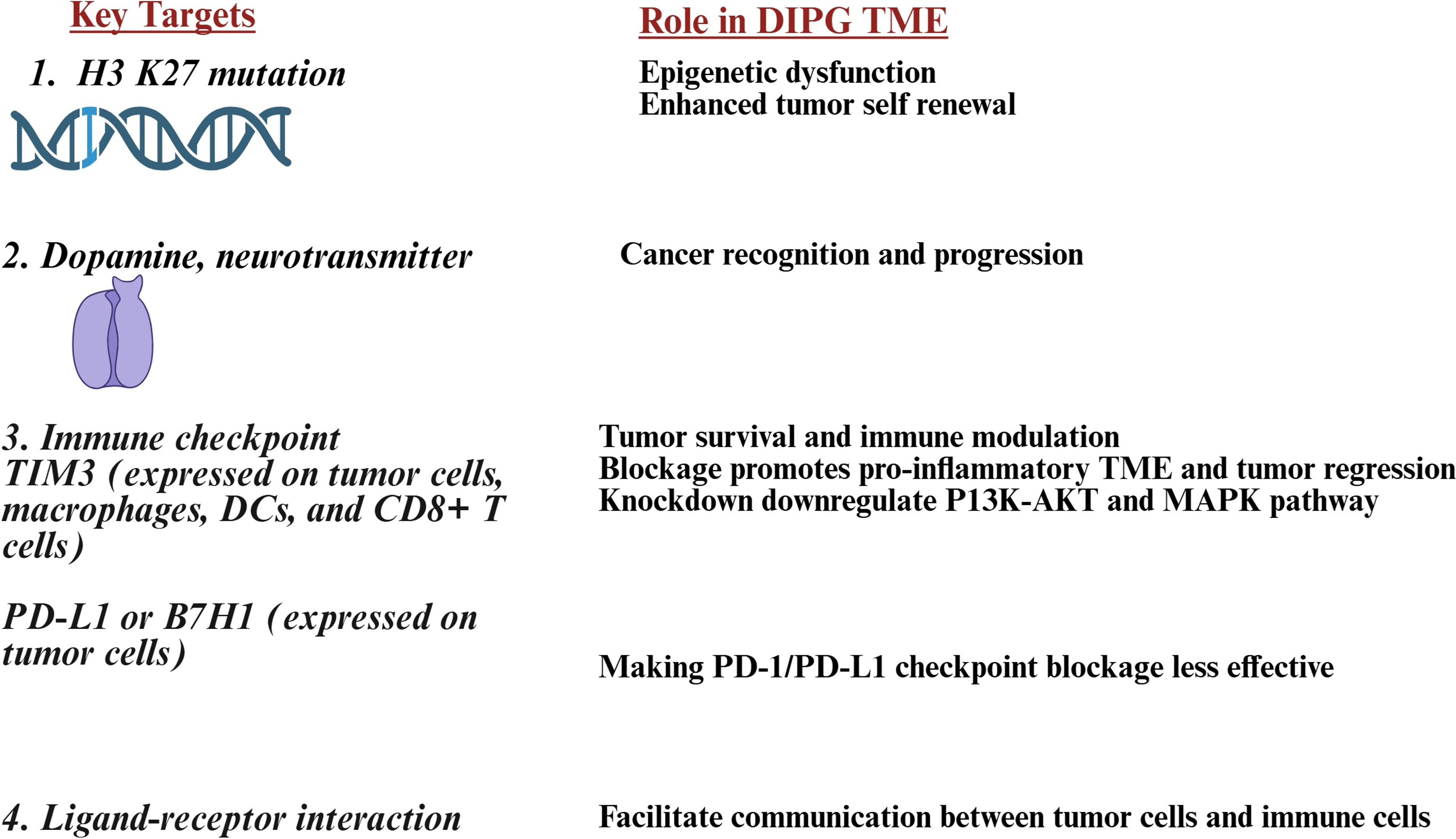
Figure 1: Illustration showing Key targets of diffuse intrinsic pontine glioma (DIPG) and their role in the tumor microenvironment (TME), these are genetic alterations, neurotransmitters, immune checkpoints, and ligand-receptor interactions. Abb: H3K27M: histone 3 K27M; TIM3: T-cell immunoglobulin and mucin-domain containing 3; DCs: dendritic cells; P13K-AKT: phosphatidylinositol 3-kinase Akt; MAPK: mitogen-activated protein kinase; PD-L1: programmed death ligand 1; B7H1: B7 homolog 1; PD-1: programmed cell death protein 1. Created in BioRender. Kaur K https://BioRender.com/1372szq (accessed on 17 November 2025)
Treatments for H3K27M-mutant diffuse midline gliomas include histone deacetylase and demethylase inhibitors, bromodomain inhibitors to block oncogenic transcription, and vaccines targeting the H3K27M neoepitope to activate the immune system [10,40,41]. Several epigenetic compounds have shown effectiveness and specificity in preclinical studies [42]. H3K27M peptide vaccines have strong preclinical evidence for immunogenicity and anti-tumor effects, and are in early-stage development [40,43]. Epigenetic therapies work by restoring balance, either by increasing H3K27me3 levels with demethylase inhibitors, reducing acetylation-driven transcription with histone deacetylase (HDAC) and bromodomain and extraterminal (BET) inhibitors, or targeting mutant epitopes through immunotherapy. Inhibiting Jumonji domain-containing protein-3 (JMJD3) demethylase with β-Alanine, N-[2-(2-pyridinyl)-6-(1,2,4,5-tetrahydro-3H-3-benzazepin-3-yl)-4-pyrimidinyl] ethylester (GSK-J4) restores K27 methylation, delays tumor progression, and extends survival in DIPG models, though its clinical development is limited due to conversion to N-[2-(2-Pyridinyl)-6-(1,2,4,5-tetrahydro-3H-3-benzazepin-3-yl)-4-pyrimidinyl]-beta-alanine (GSK-J1), which has poor permeability. A stable GSK-J1 analog developed by Suri et al. showed better anti-tumor activity and survival benefits in DIPG models [28]. Panobinostat, a pan-HDAC inhibitor, caused significant cytotoxicity in DIPG in vitro [44]. BET inhibitors have also improved survival in mouse models of H3K27-altered gliomas, with clinical trials ongoing [45,46]. Additionally, the indoleamine-2,3-dioxygenase 1 IDO1 inhibitor indoximod has shown early clinical promise against H3K27M-mutant gliomas [6,47]. This multifaceted approach offers hope for improving outcomes in this fatal pediatric brain cancer, with preclinical studies supporting these treatments individually and in combination, while clinical trials assess their safety, dosing, and efficacy [10].
2.2 Targeting Neurotransmitter
Dopamine, a neurotransmitter, plays a key role in cancer initiation and progression [48]. Dopamine receptor antagonists ONC201 and ONC206 have shown cytotoxic effects against DIPG and other diffuse midline gliomas (DMGs) with histone H3.3K27M mutations [13,49,50]. Both pediatric and adult brain tumor patients have experienced significant clinical responses to these drugs [50]. ONC201 and ONC206, part of the imipramine family of anti-cancer drugs, were discovered as a selective antagonist of the GPCR dopamine receptor D2 (DRD2) [51]. ONC201, initially identified as TIC10, induces TNF-related apoptosis and shares a similar anti-cancer mechanism as ONC206 [51,52]. These drugs have demonstrated cytotoxicity against various cancer cell lines, can cross the BBB via oral administration to target high-grade glioma, and are in clinical trials for treating central nervous system cancers [53,54]. ONC201 also activates the APO-2 pathway in cancer cells, with increased levels of APO2-L (TRAIL)-secreting NK cells observed in the peripheral blood of treated mice [55] and human patients [56].
2.3 Radiation, Chemotherapy, and Combination Therapeutics
Radiation therapy, typically fractionated external beam RT at doses of 54–59 Gy, remains the standard treatment for DIPG [57]. Re-irradiation in progressive cases may slightly extend survival, but radiation is primarily palliative and doesn’t provide long-term tumor control [57,58]. Epidermal growth factor receptor (EGFR) inhibitors like nimotuzumab, when used with radiation, can reduce side effects but haven’t significantly improved survival rates [59]. Most systemic chemotherapy regimens have been ineffective, likely due to challenges with drug delivery and tumor heterogeneity [31,32]. Additionally, DIPG shows resistance to chemotherapeutics like temozolomide [60]. Trials with kinase inhibitors like ribociclib (a CDK4/6 inhibitor) and everolimus (a mammalian target of rapamycin (mTOR) inhibitor) demonstrate safety but unclear effectiveness [61,62]. Similarly, studies on poly ADP ribose polymerase (PARP) inhibitors (e.g., veliparib), alkylating agents (temozolomide, capecitabine), topoisomerase inhibitors (irinotecan), and microtubule inhibitors (cabazitaxel) combined with radiation or chemotherapy or as adjuvants have not shown notable survival improvements [63,64].
Immunotherapy, including immune checkpoint inhibitors, CAR-T, T-cell receptor (TCR-T), vaccines, oncolytic virus, and combination therapies, has become a growing focus due to the poor outcomes and molecular features of DIPG. Understanding the tumor-immune microenvironment opens new doors for immunotherapy strategies [65] (Table 1). However, the highly immunosuppressive and low-inflammatory tumor microenvironment in DIPG limits immunotherapy effectiveness. Immune checkpoint inhibitor therapies like programmed death protein-1 (PD-1) and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) have shown good tolerance in DIPG patients [58,66,67]. However, immune checkpoint blockade therapies, such as anti-PD1, have largely failed to improve survival, likely due to the “immune-cold” tumor microenvironment with low PD-1 expression [58]. DIPG tumors typically show minimal immune cell infiltration, and infiltrated immune cells often lose PD-1 or PD-L1 expression, limiting the efficacy of immune checkpoint inhibitors [16,34]. Targeting alternative checkpoints like T cell immunoglobulin and mucin domain-containing protein 3 (TIM3) has shown promising preclinical results [68,69]. Peptide vaccines have demonstrated efficacy in inducing cytotoxic T-cell and helper-1-cell mediated responses in K27M-mutated MHC-humanized mice [70,71]. CAR-T cells, particularly anti-GD2 CAR-T cells, have shown safety and initial clinical benefits [4,72–74]. Other CAR-T approaches targeting B7-H3, HER2, and H3K27M mutant peptides are in early clinical trials, showing some encouraging tumor responses [75,76]. Autologous dendritic cell vaccines have demonstrated safety, feasibility, and anti-tumor immune responses in DIPG patients [77,78]. Cancer vaccines targeting WT1, survivin, and H3K27M antigens are under investigation to trigger tumor-specific immune reactions [79,80]. Immunomodulatory drugs like pomalidomide and pelareorep (an oncolytic virus) are being studied alone or with radiation. The efficacy of H3.3K27M-specific T-cell receptors and CAR-T was shown against DIPG in xenograft mouse models [81,82]. Positive outcomes were observed in DIPG patients treated with the immune-modulating antibody MDV9300 (pidilizumab) [83].
2.5 Therapeutic Challenges and Advancements
Treating DIPG is challenging due to its difficult location, complex molecular and epigenetic features, suppressive tumor environment, and the obstacle of getting drugs through the BBB. While innovative methods and promising models offer hope, translating them into actual survival improvements remains a big hurdle. Future strategies should prioritize personalized, multimodal treatments that penetrate the BBB, combined with targeted immunomodulation and epigenetic approaches to overcome DIPG’s resistance.
Oncolytic viral therapy, such as DNX-2401 delivered via CED and combined with radiation, has shown potential survival benefits [31,84,85]. Super selective intra-arterial cerebral infusion (SIACI) uses BBB disruption agents like mannitol with targeted antibodies such as bevacizumab (anti-VEGF) or cetuximab (anti-EGFR), showing a median overall survival (OS) of about 17.3 months in heavily pretreated patients, demonstrating safety and potential effectiveness [86]. The growing use of stereotactic biopsy aids molecular diagnosis and supports precision medicine [87]. Combination therapies involving radiation, targeted agents, immunotherapy, and BBB bypass are under active investigation [88]. Continued multidisciplinary research on genetics, drug delivery, and immune modulation is crucial to improving outcomes for this devastating pediatric cancer.
3 Natural Killer Cells in DIPG TME
BBB restricts naïve immune cells, including NK cells, to the brain tissue. DIPG creates a complex and challenging tumor immune environment. NK cells, which are vital for innate immunity and have strong tumor-killing abilities, face numerous obstacles within the DIPG TME. Studies using human data, patient-derived models, and translational work highlight these mechanisms. H3K27M-driven chromatin changes in DIPG alter cell surface ligand expression and cytokine secretion, limiting NK cell recognition and activation [89]. miRNAs in both NK and tumor cells regulate receptor expression, signaling pathways, and effector functions, enhancing NK suppression [90].
NK cell activation depends on NK activating receptors, including NKG2D, NKp30, NKp44, NKp46, CD16, and CD94, etc. [91]. These receptors detect stress ligands on tumor cells. DIPG cells and TME components express ligands for inhibitory NK receptors, such as classical major histocompatibility complex (MHC) class I interacting with KIRs, or HLA-E engaging NKG2A [91]. This engagement activates intracellular immunoreceptor tyrosine-based inhibitory motifs (ITIMs), leading to suppression of activation cascades [35]. A high inhibitory-to-activating signal ratio reduces NK cell cytotoxicity, even if tumor cells are recognized [91]. DIPG stem-like cells evade NK attacks through differences in stress ligand expression (e.g., MICA/B, RAET1) and by metabolically impairing NK cytotoxicity. Furthermore, hypoxia and nutrient competition in the TME weaken NK cell metabolic fitness and the ability to mobilize cytotoxic granules [92]. DIPG TME exhibits a low level of cytokines, including IL-15; these cytokines are crucial for NK maturation and cytotoxic activity, lack of these functions impairs NK cell-mediated tumor lysis and differentiation. Inhibitory cytokines in DIPG TME may dampen NK cell function by reducing granule polarization and degranulation efficiency [34]. Elevated B cells may inhibit NK activation indirectly by secreting IL-10 and reducing dendritic cell priming [35]. Low levels of NK-stimulating myeloid cells contribute to NK cell hyporesponsiveness [34]. Low levels of NK cell recruiting chemokines (e.g., CXCL9, CXCL10) limit the homing of peripheral NK cells to the tumor site, ultimately reducing tumor NK cell infiltration [35]. Surviving NK cells in DIPG TME present decreased cytotoxic granule release (perforin, granzyme B) and diminished cytokine secretion capability (IFN-γ, TNF-α), impairing both direct tumor lysis and immune modulation [34].
In short, NK cells are inhibited in DIPG immunosuppression TME, and contributing factors are poor immune cell recruitment, dominant inhibitory receptors, insufficient cytokine activation, and microenvironmental or metabolic challenges (Fig. 2). Effective immunotherapy strategies need to address the recruitment, activation, and sustained presence of NK cells in this immune-resistant TME.
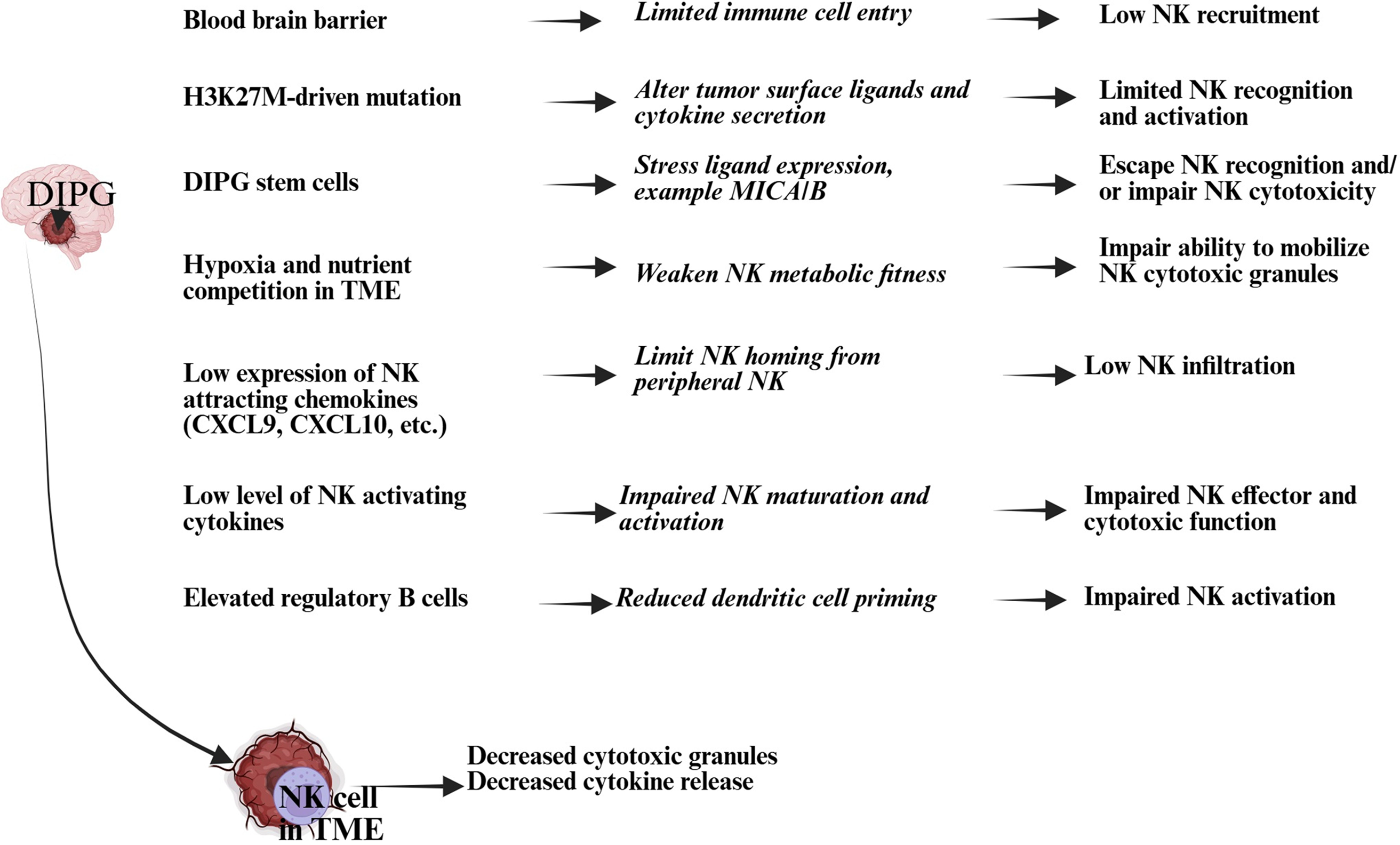
Figure 2: An illustration showing how natural killer (NK) cell activity is suppressed in the DIPG TME. NK cell activity plays a vital role in tumor destruction and growth control; reduced NK cell numbers and functionality lead to tumor progression and spread. Created in BioRender. Kaur K https://BioRender.com/9g38tjj (accessed on 17 November 2025). Abb: CXCL: Chemokine (C-X-X motif) ligand; MICA/B: major histocompatibility complex (MHC) class I chain-related protein A/B
4 NK Cell-Based Immunotherapies for DIPG: Preclinical and Clinical Studies
NK cells have shown the ability to destroy DIPG tumor cells in vitro [93,94] (Table 2 and Fig. 3). They can selectively recognize and kill DIPG tumor cells, including glioma stem cells, overcoming some tumor immune evasion mechanisms, suggesting their therapeutic promise [95]. Preclinical studies convincingly demonstrate that NK cells, especially when engineered as CAR-NK cells or engaged via bispecific molecules, offer promising targeted immunotherapeutic approaches for DIPG [96–99]. CAR-NK cells exhibit transient viability and cytotoxicity following irradiation (a safety step before infusion), highlighting their translational potential [100]. CAR-engineered NK (CAR-NK) cells targeting tumor-associated antigens like GD2 have shown strong, antigen-specific cytotoxicity against DIPG cells with high GD2 expression in vitro and in orthotopic xenograft models, resulting in tumor growth inhibition and improved survival in preclinical mouse studies [100,101]. In mouse models, GD2-CAR NK-92 cells slowed tumor growth and extended survival in GD2-positive DIPG xenografts with an acceptable safety profile. GD2-CAR NK-92 cell lines specifically kill GD2-expressing DIPG cells with minimal toxicity to normal neural progenitor cells and show favorable safety profiles in vivo, avoiding cytokine release syndrome in treated mice [102]. Despite challenges posed by the DIPG tumor microenvironment and blood-brain barrier, these strategies have shown effective tumor control and survival benefits in animal models, with favorable safety profiles [16,17]. Future trials will need to confirm the preclinical efficacy and safety of CAR-NK cell therapies, such as GD2-CAR NK cells, before they reach clinical stages. The adoptive transfer of irradiated CAR-NK-92 cells in preclinical models shows a gradual loss of viability and cytotoxicity over time, raising challenges for in vivo persistence [103].

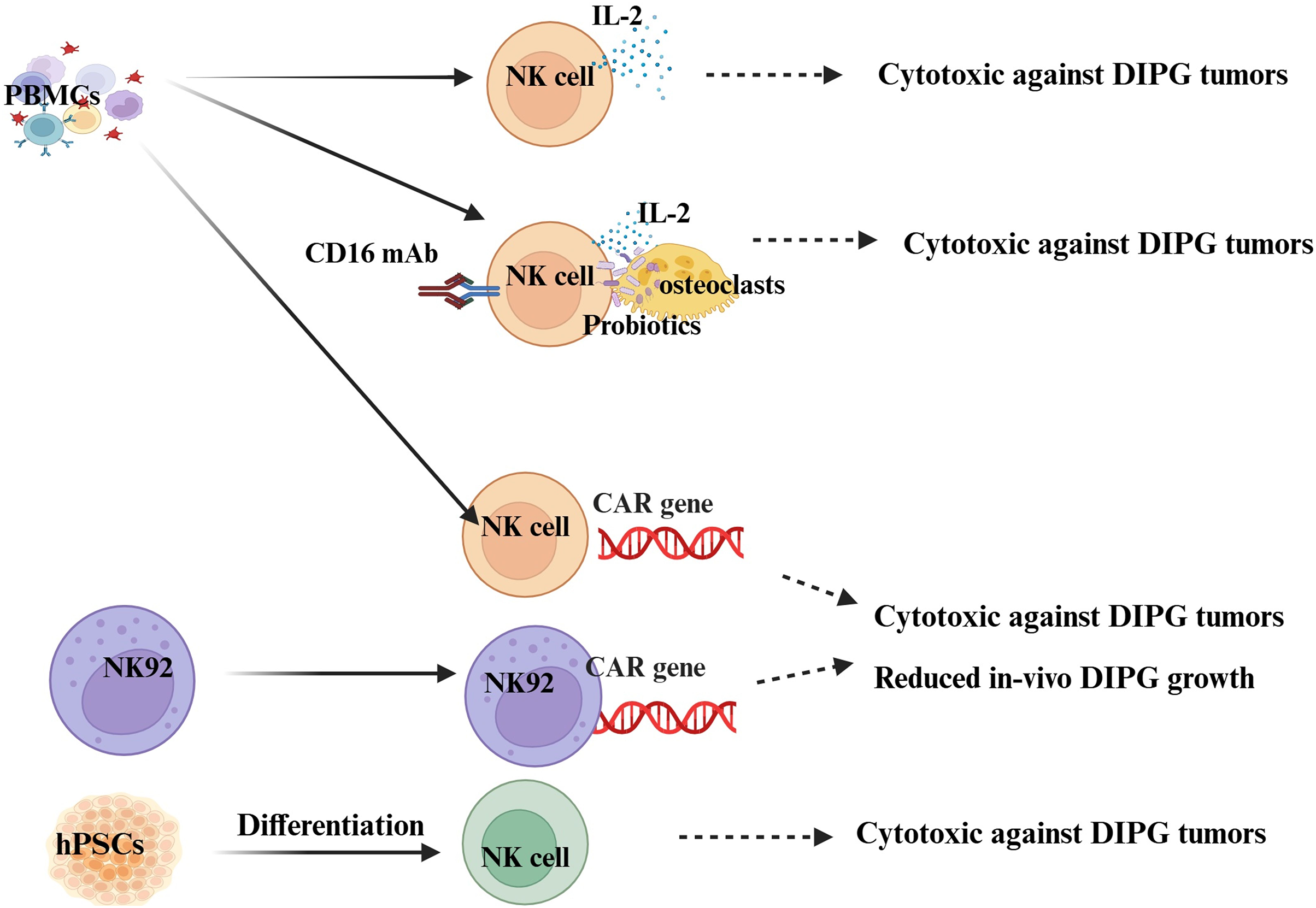
Figure 3: Illustration showcasing NK cell therapy platforms for DIPG. This can be achieved through cytokines, a combination of feeder cells, cytokines, Fc antibodies, and probiotics, or by utilizing CAR technology. Created in BioRender. Kaur et al. https://BioRender.com/rinjnt8 (accessed on 17 November 2025). PBMCs: peripheral blood mononuclear cells; CD16: cluster of differentiation 16; hPSCs: human pluripotent stem cells; CAR: chimeric antigen receptor
Beyond CAR-NK cells, innovative therapies like NK cell Engagers (NKCEs) are being developed to bind NK cells and tumor antigens simultaneously, and have the potential to boost NK cell recruitment and activation against DIPG cells [104,105]. Investigational NK cell-derived exosomes and other NK-based approaches are also being explored to target glioma stem cells (GSCs), which contribute to DIPG aggressiveness and treatment resistance [106,107]. Ex vivo osteoclasts-induced expanded supercharged NK cells were found to be effective in targeting DIPG tumors alone or in combination with the dopamine receptor antagonists (ONC201 and ONC206) [93,94]. Treating NK cells with epigenetic modifiers like HDAC inhibitors (e.g., MS-275) boosts their cytotoxicity against solid cancers by enhancing genes linked to movement and cell killing [108,109].
Translation into clinical trials is underway or imminent, underscoring the potential for NK cell-based therapies to become a vital component in treating this devastating pediatric brain cancer [17,97]. Currently, no large-scale clinical trials have been published deploying NK cell therapies (unmodified or CAR-engineered) in DIPG patients. Most ongoing clinical research focuses on CAR-T cell therapies targeting antigens like GD2 and B7-H3, which have shown promising results in early to mid-phase trials (ClinicalTrials.gov. ID NCT04196413). Preliminary data suggest that CAR-NK therapies might have more favorable safety profiles and lower cytokine release syndrome (CRS) risks compared to CAR-T therapies [111,112]. CAR-NK cells offer improved tumor specificity and cytotoxicity with lower risks of graft-versus-host disease or cytokine release syndrome compared to CAR-T cells [37]. Understanding NK cell trafficking, persistence, and activation in the DIPG microenvironment is critical. The potential of combining NK cell therapy with epigenetic-modifying agents has yet to be clinically tested. There is a pressing need for clinical trials focusing on NK-cell therapies, including CAR-NK cells, in DIPG.
5 Challenges and Future Perspectives
Adoptively transferred NK cells face significant challenges with survival and proliferation in vivo, especially in the immunosuppressive TME [89]. Physical barriers and low chemokine levels can hinder NK cell recruitment and infiltration into DIPG [89]. Cytokine therapies like IL-2-activated NK cells often cause severe toxicities, such as vascular leak syndrome and cytokine release syndrome [113,114]. Tumors evade NK cell function by downregulating activating ligands or releasing soluble ligands like MICA/B, which interact with inhibitory NK receptors and lead to dysfunction [92]. They may also maintain or increase MHC class I molecule expression, activating inhibitory KIRs on NK cells [35]. Inhibitory checkpoint molecules like PD-1 contribute to NK cell exhaustion and reduced cytotoxicity, while factors like TGF-β, IL-10, and hypoxia further suppress NK cell survival and activation [115–118]. Advanced strategies aim to create memory-like NK cells, such as cytokine-induced memory-like NK cells, to enhance their persistence and antitumor efficacy.
Allogeneic NK cells show great potential for adoptive therapies because they carry a low risk of GVHD, but they face hurdles like immune rejection and limited persistence due to HLA mismatches [119,120]. Functional immune systems in recipients often recognize and reject these foreign cells [39]. Host T cells can attack donor cells with mismatched HLA, B cells may produce alloantibodies tagging NK cells for destruction, and host NK cells might target donor cells lacking self-HLA ligands [121,122]. Additionally, macrophages and complement activation contribute to the elimination of donor NK cells, limiting their effectiveness [123]. Selecting donors based on KIR and HLA compatibility can boost NK cell function and reduce rejection, while repeated doses of off-the-shelf NK products may improve therapeutic outcomes [124].
NK cells have shown promise in preclinical studies against DIPG, both naturally and when CAR-engineered. Studies have demonstrated safety and effectiveness in lab and animal models, particularly with GD2-CAR NK-92 cells targeting GD2-positive DIPG cells. However, clinical evidence for NK cell therapies in DIPG remains limited, with no late-phase trials yet, unlike CAR-T therapies. Challenges include the tumor’s immunosuppressive environment, limited NK cell persistence after infusion, and determining the best delivery methods—whether intratumoral, intraventricular, or systemic. Enhancing NK cell function through epigenetic priming or combination therapies is a promising research area. Moving forward, priorities include clinical trials on NK cell therapies, identifying biomarkers for treatment response, and ensuring patient safety. The progress of CAR-T cells provides a strong foundation to advance CAR-NK therapies into early-phase trials.
Acknowledgement: None.
Funding Statement: The author received no specific funding for this study.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The author declares no conflicts of interest to report regarding the present study.
References
1. Venkatesh HS, Tam LT, Woo PJ, Lennon J, Nagaraja S, Gillespie SM, et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature. 2017;549(7673):533–7. doi:10.1038/nature24014. [Google Scholar] [PubMed] [CrossRef]
2. Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell. 2015;161(4):803–16. doi:10.1016/j.cell.2015.04.012. [Google Scholar] [PubMed] [CrossRef]
3. Mandorino M, Maitra A, Armenise D, Baldelli OM, Miciaccia M, Ferorelli S, et al. Pediatric diffuse midline glioma H3K27-altered: from developmental origins to therapeutic challenges. Cancers. 2024;16(10):1814. doi:10.3390/cancers16101814. [Google Scholar] [PubMed] [CrossRef]
4. Vitanza NA, Monje M. Diffuse intrinsic pontine glioma: from diagnosis to next-generation clinical trials. Curr Treat Options Neurol. 2019;21(8):37. doi:10.1007/s11940-019-0577-y. [Google Scholar] [PubMed] [CrossRef]
5. Rashed WM, Maher E, Adel M, Saber O, Zaghloul MS. Pediatric diffuse intrinsic pontine glioma: where do we stand? Cancer Metastasis Rev. 2019;38(4):759–70. doi:10.1007/s10555-019-09824-2. [Google Scholar] [PubMed] [CrossRef]
6. Wierzbicki K, Ravi K, Franson A, Bruzek A, Cantor E, Harris M, et al. Targeting and therapeutic monitoring of H3K27M-mutant glioma. Curr Oncol Rep. 2020;22(2):19. doi:10.1007/s11912-020-0877-0. [Google Scholar] [PubMed] [CrossRef]
7. Harutyunyan AS, Krug B, Chen H, Papillon-Cavanagh S, Zeinieh M, De Jay N, et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat Commun. 2019;10(1):1262. doi:10.1038/s41467-019-09140-x. [Google Scholar] [PubMed] [CrossRef]
8. Saratsis AM, Knowles T, Petrovic A, Nazarian J. H3K27M mutant glioma: disease definition and biological underpinnings. Neuro Oncol. 2024;26(Suppl_2):S92–100. doi:10.1093/neuonc/noad164. [Google Scholar] [PubMed] [CrossRef]
9. Konuma T, Zhou MM. Distinct histone H3 lysine 27 modifications dictate different outcomes of gene transcription. J Mol Biol. 2024;436(7):168376. doi:10.1016/j.jmb.2023.168376. [Google Scholar] [PubMed] [CrossRef]
10. Yang Z, Sun L, Chen H, Sun C, Xia L. New progress in the treatment of diffuse midline glioma with H3K27M alteration. Heliyon. 2024;10(2):e24877. doi:10.1016/j.heliyon.2024.e24877. [Google Scholar] [PubMed] [CrossRef]
11. Arvanitis CD, Ferraro GB, Jain RK. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat Rev Cancer. 2020;20(1):26–41. doi:10.1038/s41568-019-0205-x. [Google Scholar] [PubMed] [CrossRef]
12. Damodharan S, Lara-Velazquez M, Williamsen BC, Helgager J, Dey M. Diffuse intrinsic pontine glioma: molecular landscape, evolving treatment strategies and emerging clinical trials. J Pers Med. 2022;12(5):840. doi:10.3390/jpm12050840. [Google Scholar] [PubMed] [CrossRef]
13. Perrone MG, Ruggiero A, Centonze A, Carrieri A, Ferorelli S, Scilimati A. Diffuse intrinsic pontine glioma (DIPGbreakthrough and clinical perspective. Curr Med Chem. 2021;28(17):3287–317. doi:10.2174/0929867327666200806110206. [Google Scholar] [PubMed] [CrossRef]
14. Frazier JL, Lee J, Thomale UW, Noggle JC, Cohen KJ, Jallo GI. Treatment of diffuse intrinsic brainstem gliomas: failed approaches and future strategies. J Neurosurg Pediatr. 2009;3(4):259–69. doi:10.3171/2008.11.PEDS08281. [Google Scholar] [PubMed] [CrossRef]
15. Bentayebi K, El Aked R, Ezzahidi O, Alami AB, Louati S, Ouadghiri M, et al. Targeting molecular mechanisms underlying treatment efficacy and resistance in DIPG: a review of current and future strategies. Brain Disord. 2024;14:100132. doi:10.1016/j.dscb.2024.100132. [Google Scholar] [CrossRef]
16. Weisbrod LJ, Thiraviyam A, Vengoji R, Shonka N, Jain M, Ho W, et al. Diffuse intrinsic pontine glioma (DIPGa review of current and emerging treatment strategies. Cancer Lett. 2024;590:216876. doi:10.1016/j.canlet.2024.216876. [Google Scholar] [PubMed] [CrossRef]
17. Fares J, Davis ZB, Rechberger JS, Toll SA, Schwartz JD, Daniels DJ, et al. Advances in NK cell therapy for brain tumors. npj Precis Onc. 2023;7(1):17. doi:10.1038/s41698-023-00356-1. [Google Scholar] [PubMed] [CrossRef]
18. Pan C, Zhai Y, Li G, Jiang T, Zhang W. NK cell-based immunotherapy and therapeutic perspective in gliomas. Front Oncol. 2021;11:751183. doi:10.3389/fonc.2021.751183. [Google Scholar] [PubMed] [CrossRef]
19. Tseng HC, Inagaki A, Bui VT, Cacalano N, Kasahara N, Man YG, et al. Differential targeting of stem cells and differentiated glioblastomas by NK cells. J Cancer. 2015;6(9):866–76. doi:10.7150/jca.11527. [Google Scholar] [PubMed] [CrossRef]
20. Kozlowska AK, Tseng HC, Kaur K, Topchyan P, Inagaki A, Bui VT, et al. Resistance to cytotoxicity and sustained release of interleukin-6 and interleukin-8 in the presence of decreased interferon-γ after differentiation of glioblastoma by human natural killer cells. Cancer Immunol Immunother. 2016;65(9):1085–97. doi:10.1007/s00262-016-1866-x. [Google Scholar] [PubMed] [CrossRef]
21. Breznik B, Ko MW, Tse C, Chen PC, Senjor E, Majc B, et al. Infiltrating natural killer cells bind, lyse and increase chemotherapy efficacy in glioblastoma stem-like tumorospheres. Commun Biol. 2022;5(1):436. doi:10.1038/s42003-022-03402-z. [Google Scholar] [PubMed] [CrossRef]
22. Daher M, Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr Opin Immunol. 2018;51:146–53. doi:10.1016/j.coi.2018.03.013. [Google Scholar] [PubMed] [CrossRef]
23. Palmer JM, Rajasekaran K, Thakar MS, Malarkannan S. Clinical relevance of natural killer cells following hematopoietic stem cell transplantation. J Cancer. 2013;4(1):25–35. doi:10.7150/jca.5049. [Google Scholar] [PubMed] [CrossRef]
24. Fildes JE, Yonan N, Leonard CT. Natural killer cells and lung transplantation, roles in rejection, infection, and tolerance. Transpl Immunol. 2008;19(1):1–11. doi:10.1016/j.trim.2008.01.004. [Google Scholar] [PubMed] [CrossRef]
25. Farag SS, Caligiuri MA. Human natural killer cell development and biology. Blood Rev. 2006;20(3):123–37. doi:10.1016/j.blre.2005.10.001. [Google Scholar] [PubMed] [CrossRef]
26. Avril T, Vauleon E, Hamlat A, Saikali S, Etcheverry A, Delmas C, et al. Human glioblastoma stem-like cells are more sensitive to allogeneic NK and T cell-mediated killing compared with serum-cultured glioblastoma cells. Brain Pathol. 2012;22(2):159–74. doi:10.1111/j.1750-3639.2011.00515.x. [Google Scholar] [PubMed] [CrossRef]
27. Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A, et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol. 2009;182(6):3530–9. doi:10.4049/jimmunol.0802845. [Google Scholar] [PubMed] [CrossRef]
28. Poli A, Wang J, Domingues O, Planagumà J, Yan T, Rygh CB, et al. Targeting glioblastoma with NK cells and mAb against NG2/CSPG4 prolongs animal survival. Oncotarget. 2013;4(9):1527–46. doi:10.18632/oncotarget.1291. [Google Scholar] [PubMed] [CrossRef]
29. Jewett A, Tseng HC. Tumor induced inactivation of natural killer cell cytotoxic function; implication in growth, expansion and differentiation of cancer stem cells. J Cancer. 2011;2:443–57. doi:10.7150/jca.2.443. [Google Scholar] [PubMed] [CrossRef]
30. Kaur K, Cook J, Park SH, Topchyan P, Kozlowska A, Ohanian N, et al. Novel strategy to expand super-charged NK cells with significant potential to lyse and differentiate cancer stem cells: differences in NK expansion and function between healthy and cancer patients. Front Immunol. 2017;8(4):297. doi:10.3389/fimmu.2017.00297. [Google Scholar] [PubMed] [CrossRef]
31. El-Khouly FE, van Vuurden DG, Stroink T, Hulleman E, Kaspers GJL, Hendrikse NH, et al. Effective drug delivery in diffuse intrinsic pontine glioma: a theoretical model to identify potential candidates. Front Oncol. 2017;7:254. doi:10.3389/fonc.2017.00254. [Google Scholar] [PubMed] [CrossRef]
32. Arms LM, Duchatel RJ, Jackson ER, Sobrinho PG, Dun MD, Hua S. Current status and advances to improving drug delivery in diffuse intrinsic pontine glioma. J Control Release. 2024;370(23):835–65. doi:10.1016/j.jconrel.2024.05.018. [Google Scholar] [PubMed] [CrossRef]
33. Tykocki T. Diffuse intrinsic pontine glioma and chimeric antigen receptor T-cell therapy: an emerging frontier. World Neurosurg. 2025;194(Suppl 1):123579. doi:10.1016/j.wneu.2024.123579. [Google Scholar] [PubMed] [CrossRef]
34. den Ende B Van, Riva M, De Smet F, Jacobs S, Hulleman E, Coosemans A. Exploring the tumor microenvironment in diffuse intrinsic pontine glioma: immunological insights and therapeutic challenges. J Immunother Cancer. 2025;13(6):e012009. doi:10.1136/jitc-2025-012009. [Google Scholar] [PubMed] [CrossRef]
35. Zhang L, Yu H, Xue Y, Liu Y. Decreased natural killer cells in diffuse intrinsic pontine glioma patients. Childs Nerv Syst. 2020;36(7):1345–6. doi:10.1007/s00381-020-04665-9. [Google Scholar] [PubMed] [CrossRef]
36. Li Y, Rezvani K, Rafei H. Next-generation chimeric antigen receptors for T- and natural killer-cell therapies against cancer. Immunol Rev. 2023;320(1):217–35. doi:10.1111/imr.13255. [Google Scholar] [PubMed] [CrossRef]
37. Peng L, Sferruzza G, Yang L, Zhou L, Chen S. CAR-T and CAR-NK as cellular cancer immunotherapy for solid tumors. Cell Mol Immunol. 2024;21(10):1089–108. doi:10.1038/s41423-024-01207-0. [Google Scholar] [PubMed] [CrossRef]
38. Yang R, Yang Y, Liu R, Wang Y, Yang R, He A. Advances in CAR-NK cell therapy for hematological malignancies. Front Immunol. 2024;15:1414264. doi:10.3389/fimmu.2024.1414264. [Google Scholar] [PubMed] [CrossRef]
39. Berrien-Elliott MM, Jacobs MT, Fehniger TA. Allogeneic natural killer cell therapy. Blood. 2023;141(8):856–68. doi:10.1182/blood.2022016200. [Google Scholar] [PubMed] [CrossRef]
40. Grassl N, Poschke I, Lindner K, Bunse L, Mildenberger I, Boschert T, et al. A H3K27M-targeted vaccine in adults with diffuse midline glioma. Nat Med. 2023;29(10):2586–92. doi:10.1038/s41591-023-02555-6. [Google Scholar] [PubMed] [CrossRef]
41. Lo Greco MC, Marano G, La Rocca M, Acquaviva G, Milazzotto R, Liardo RLE, et al. Latest advancements in the management of H3K27M-mutant diffuse intrinsic pontine glioma: a narrative review. Cancers. 2025;17(3):420. doi:10.3390/cancers17030420. [Google Scholar] [PubMed] [CrossRef]
42. Viaene AN. Pediatric brain tumors: a neuropathologist’s approach to the integrated diagnosis. Front Pediatr. 2023;11:1143363. doi:10.3389/fped.2023.1143363. [Google Scholar] [PubMed] [CrossRef]
43. Boschert T, Kromer K, Lerner T, Lindner K, Haltenhof G, Tan CL, et al. H3K27M neoepitope vaccination in diffuse midline glioma induces B and T cell responses across diverse HLA loci of a recovered patient. Sci Adv. 2024;10(5):1–11. doi:10.1126/sciadv.adi9091. [Google Scholar] [PubMed] [CrossRef]
44. Bentayebi K, Suwan K, Ibrahimi A, Sara L, Ouadghiri M, Aanniz T, et al. Preclinical evaluation of panobinostat and ONC201 for the treatment of diffuse intrinsic pontine glioma (DIPG). Brain Disord. 2024;13:100113. doi:10.1016/j.dscb.2023.100113. [Google Scholar] [CrossRef]
45. Watanabe J, Clutter MR, Gullette MJ, Sasaki T, Uchida E, Kaur S, et al. BET bromodomain inhibition potentiates radiosensitivity in models of H3K27-altered diffuse midline glioma. J Clin Invest. 2024;134(13):e174794. doi:10.1172/JCI174794. [Google Scholar] [PubMed] [CrossRef]
46. Shorstova T, Foulkes WD, Witcher M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br J Cancer. 2021;124(9):1478–90. doi:10.1038/s41416-021-01321-0. [Google Scholar] [PubMed] [CrossRef]
47. Gao M, Yu W, Xi Z, Zhang Z, Fan X, Wang X. Recent update on the discovery of indoleamine-2,3-dioxygenase 1 inhibitors targeting cancer immunotherapy. Eur J Med Chem. 2025;298:118017. doi:10.1016/j.ejmech.2025.118017. [Google Scholar] [PubMed] [CrossRef]
48. Caragher SP, Hall RR, Ahsan R, Ahmed AU. Monoamines in glioblastoma: complex biology with therapeutic potential. Neuro Oncol. 2018;20(8):1014–25. doi:10.1093/neuonc/nox210. [Google Scholar] [PubMed] [CrossRef]
49. Tu YS, He J, Liu H, Davis RE, Orlowski RZ, Allen JE, et al. ONC201 overcomes chemotherapy resistance by upregulation of BIM in multiple myeloma. Blood. 2016;128(22):4476. doi:10.1182/blood.v128.22.4476.4476. [Google Scholar] [CrossRef]
50. Bonner ER, Waszak SM, Grotzer MA, Mueller S, Nazarian J. Mechanisms of imipridones in targeting mitochondrial metabolism in cancer cells. Neuro Oncol. 2021;23(4):542–56. doi:10.1093/neuonc/noaa283. [Google Scholar] [PubMed] [CrossRef]
51. Prabhu VV, Morrow S, Rahman Kawakibi A, Zhou L, Ralff M, Ray J et al. ONC201 and imipridones: anti-cancer compounds with clinical efficacy. Neoplasia. 2020;22(12):725–44. doi:10.1016/j.neo.2020.09.005. [Google Scholar] [PubMed] [CrossRef]
52. Pathak SO, Manohar SM. ONC201 (dordaviprone) induces integrated stress response and death in cervical cancer cells. Biomolecules. 2025;15(4):463. doi:10.3390/biom15040463. [Google Scholar] [PubMed] [CrossRef]
53. El-Soussi S, Hanna R, Semaan H, Khater AR, Abdallah J, Abou-Kheir W, et al. A novel therapeutic mechanism of imipridones ONC201/ONC206 in MYCN-amplified neuroblastoma cells via differential expression of tumorigenic proteins. Front Pediatr. 2021;9:693145. doi:10.3389/fped.2021.693145. [Google Scholar] [PubMed] [CrossRef]
54. Zhou L, Zhang L, Zhang J, Wu LJ, Zhang S, George A, et al. Imipridones ONC201/ONC206 + RT/TMZ triple (IRT) therapy reduces intracranial tumor burden, prolongs survival in orthotopic IDH-WT GBM mouse model, and suppresses MGMT. Oncotarget. 2025;16(1):230–48. doi:10.18632/oncotarget.28707. [Google Scholar] [PubMed] [CrossRef]
55. Wu LJ, Pinho-Schwermann M, Zhou L, Zhang L, Huntington KE, Malpass R, et al. Synergistic combination therapy with ONC201 or ONC206, and enzalutamide or darolutamide in preclinical studies of castration-resistant prostate cancer. Am J Cancer Res. 2024;14(12):6012–36. doi:10.62347/VJMW4904. [Google Scholar] [PubMed] [CrossRef]
56. Wagner J, Kline CL, Zhou L, Campbell KS, MacFarlane AW, Olszanski AJ, et al. Dose intensification of TRAIL-inducing ONC201 inhibits metastasis and promotes intratumoral NK cell recruitment. J Clin Invest. 2018;128(6):2325–38. doi:10.1172/JCI96711. [Google Scholar] [PubMed] [CrossRef]
57. Gallitto M, Lazarev S, Wasserman I, Stafford JM, Wolden SL, Terezakis SA, et al. Role of radiation therapy in the management of diffuse intrinsic pontine glioma: a systematic review. Adv Radiat Oncol. 2019;4(3):520–31. doi:10.1016/j.adro.2019.03.009. [Google Scholar] [PubMed] [CrossRef]
58. Kline C, Liu SJ, Duriseti S, Banerjee A, Nicolaides T, Raber S, et al. Reirradiation and PD-1 inhibition with nivolumab for the treatment of recurrent diffuse intrinsic pontine glioma: a single-institution experience. J Neurooncol. 2018;140(3):629–38. doi:10.1007/s11060-018-2991-5. [Google Scholar] [PubMed] [CrossRef]
59. Bossi P, Platini F. Radiotherapy plus EGFR inhibitors: synergistic modalities. Cancers Head Neck. 2017;2(1):2. doi:10.1186/s41199-016-0020-y. [Google Scholar] [PubMed] [CrossRef]
60. Heravi Shargh V, Luckett J, Bouzinab K, Paisey S, Turyanska L, Singleton WGB, et al. Chemosensitization of temozolomide-resistant pediatric diffuse midline glioma using potent nanoencapsulated forms of a N(3)-propargyl analogue. ACS Appl Mater Interfaces. 2021;13(30):35266–80. doi:10.1021/acsami.1c04164. [Google Scholar] [PubMed] [CrossRef]
61. Braal CL, Jongbloed EM, Wilting SM, Mathijssen RHJ, Koolen SLW, Jager A. Inhibiting CDK4/6 in breast cancer with palbociclib, ribociclib, and abemaciclib: similarities and differences. Drugs. 2021;81(3):317–31. doi:10.1007/s40265-020-01461-2. [Google Scholar] [PubMed] [CrossRef]
62. Johnston S, Emde A, Barrios C, Srock S, Neven P, Martin M, et al. Cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors: existing and emerging differences. JNCI Cancer Spectr. 2023;7(4):1–11. doi:10.1093/jncics/pkad045. [Google Scholar] [PubMed] [CrossRef]
63. Pommier Y, Murai J. O6.1-classification of PARP inhibitors based on PARP trapping and catalytic inhibition, and rationale for combinations. Ann Oncol. 2015;26:ii8. doi:10.1093/annonc/mdv084.1. [Google Scholar] [CrossRef]
64. Bondar D, Karpichev Y. Poly(ADP-ribose) polymerase (PARP) inhibitors for cancer therapy: advances, challenges, and future directions. Biomolecules. 2024;14(10):1269. doi:10.3390/biom14101269. [Google Scholar] [PubMed] [CrossRef]
65. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–50. doi:10.1038/s41591-018-0014-x. [Google Scholar] [PubMed] [CrossRef]
66. Cacciotti C, Choi J, Alexandrescu S, Zimmerman MA, Cooney TM, Chordas C, et al. Immune checkpoint inhibition for pediatric patients with recurrent/refractory CNS tumors: a single institution experience. J Neurooncol. 2020;149(1):113–22. doi:10.1007/s11060-020-03578-6. [Google Scholar] [PubMed] [CrossRef]
67. Garg P, Pareek S, Kulkarni P, Horne D, Salgia R, Singhal SS. Next-generation immunotherapy: advancing clinical applications in cancer treatment. J Clin Med. 2024;13(21):6537. doi:10.3390/jcm13216537. [Google Scholar] [PubMed] [CrossRef]
68. Cai L, Li Y, Tan J, Xu L, Li Y. Targeting LAG-3, TIM-3, and TIGIT for cancer immunotherapy. J Hematol Oncol. 2023;16(1):101. doi:10.1186/s13045-023-01499-1. [Google Scholar] [PubMed] [CrossRef]
69. Ausejo-Mauleon I, Labiano S, de la Nava D, Laspidea V, Zalacain M, Marrodán L, et al. TIM-3 blockade in diffuse intrinsic pontine glioma models promotes tumor regression and antitumor immune memory. Cancer Cell. 2023;41(11):1911–26. doi:10.1016/j.ccell.2023.09.001. [Google Scholar] [PubMed] [CrossRef]
70. Abd-Aziz N, Poh CL. Development of peptide-based vaccines for cancer. J Oncol. 2022;2022(1):9749363. doi:10.1155/2022/9749363. [Google Scholar] [PubMed] [CrossRef]
71. Ochs K, Ott M, Bunse T, Sahm F, Bunse L, Deumelandt K, et al. K27M-mutant histone-3 as a novel target for glioma immunotherapy. OncoImmunology. 2017;6(7):e1328340. doi:10.1080/2162402X.2017.1328340. [Google Scholar] [PubMed] [CrossRef]
72. Alnefaie A, Albogami S, Asiri Y, Ahmad T, Alotaibi SS, Al-Sanea MM, et al. Chimeric antigen receptor T-cells: an overview of concepts, applications, limitations, and proposed solutions. Front Bioeng Biotechnol. 2022;10:797440. doi:10.3389/fbioe.2022.797440. [Google Scholar] [PubMed] [CrossRef]
73. Chiavelli C, Prapa M, Rovesti G, Silingardi M, Neri G, Pugliese G, et al. Autologous anti-GD2 CAR T cells efficiently target primary human glioblastoma. npj Precis Oncol. 2024;8(1):26. doi:10.1038/s41698-024-00506-z. [Google Scholar] [PubMed] [CrossRef]
74. Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat Med. 2018;24(5):572–9. doi:10.1038/s41591-018-0006-x. [Google Scholar] [PubMed] [CrossRef]
75. Vitanza NA, Ronsley R, Choe M, Seidel K, Huang W, Rawlings-Rhea SD, et al. Intracerebroventricular B7-H3-targeting CAR T cells for diffuse intrinsic pontine glioma: a phase 1 trial. Nat Med. 2025;31(3):861–8. doi:10.1038/s41591-024-03451-3. [Google Scholar] [PubMed] [CrossRef]
76. Li N, Zhang C, Li X, Liu S, Xu Y, Yang X. Targeting B7-H3 in solid tumors: development and evaluation of novel CAR-T cell therapy. Immunobiology. 2025;230(3):152888. doi:10.1016/j.imbio.2025.152888. [Google Scholar] [PubMed] [CrossRef]
77. Benitez-Ribas D, Cabezón R, Flórez-Grau G, Molero MC, Puerta P, Guillen A, et al. Immune response generated with the administration of autologous dendritic cells pulsed with an allogenic tumoral cell-lines lysate in patients with newly diagnosed diffuse intrinsic pontine glioma. Front Oncol. 2018;8:127. doi:10.3389/fonc.2018.00127. [Google Scholar] [PubMed] [CrossRef]
78. Dillman RO, Nistor GI, Keirstead HS. Autologous dendritic cells loaded with antigens from self-renewing autologous tumor cells as patient-specific therapeutic cancer vaccines. Hum Vaccin Immunother. 2023;19(1):2198467. doi:10.1080/21645515.2023.2198467. [Google Scholar] [PubMed] [CrossRef]
79. Ogasawara M. Wilms’ tumor 1-targeting cancer vaccine: recent advancements and future perspectives. Hum Vaccin Immunother. 2024;20(1):2296735. doi:10.1080/21645515.2023.2296735. [Google Scholar] [PubMed] [CrossRef]
80. Fan T, Zhang M, Yang J, Zhu Z, Cao W, Dong C. Therapeutic cancer vaccines: advancements, challenges, and prospects. Signal Transduct Target Ther. 2023;8(1):450. doi:10.1038/s41392-023-01674-3. [Google Scholar] [PubMed] [CrossRef]
81. Chheda ZS, Kohanbash G, Okada K, Jahan N, Sidney J, Pecoraro M, et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med. 2018;215(1):141–57. doi:10.1084/jem.20171046. [Google Scholar] [PubMed] [CrossRef]
82. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–90. doi:10.1038/nm.3838. [Google Scholar] [PubMed] [CrossRef]
83. Fried I, Lossos A, Ben Ami T, Dvir R, Toledano H, Ben Arush MW, et al. Preliminary results of immune modulating antibody MDV9300 (pidilizumab) treatment in children with diffuse intrinsic pontine glioma. J Neurooncol. 2018;136(1):189–95. doi:10.1007/s11060-017-2643-1. [Google Scholar] [PubMed] [CrossRef]
84. Nassiri F, Patil V, Yefet LS, Singh O, Liu J, Dang RMA, et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: a phase 1/2 trial. Nat Med. 2023;29(6):1370–8. doi:10.1038/s41591-023-02347-y. [Google Scholar] [PubMed] [CrossRef]
85. Pérez-Larraya JG, Garcia-Moure M, Labiano S, Patiño-García A, Dobbs J, Gonzalez-Huarriz M, et al. Oncolytic DNX-2401 virus for pediatric diffuse intrinsic pontine glioma. New Engl J Med. 2022;386(26):2471–81. [Google Scholar]
86. D’Amico RS, Khatri D, Reichman N, Patel NV, Wong T, Fralin SR, et al. Super selective intra-arterial cerebral infusion of modern chemotherapeutics after blood-brain barrier disruption: where are we now, and where we are going. J Neuro Oncol. 2020;147(2):261–78. doi:10.1007/s11060-020-03435-6. [Google Scholar] [PubMed] [CrossRef]
87. Katzendobler S, Do A, Weller J, Rejeski K, Dorostkar MM, Albert NL, et al. The value of stereotactic biopsy of primary and recurrent brain metastases in the era of precision medicine. Front Oncol. 2022;12:1014711. doi:10.3389/fonc.2022.1014711. [Google Scholar] [PubMed] [CrossRef]
88. Wisdom AJ, Barker CA, Chang JY, Demaria S, Formenti S, Grassberger C, et al. The next chapter in immunotherapy and radiation combination therapy: cancer-specific perspectives. Int J Radiat Oncol Biol Phys. 2024;118(5):1404–21. doi:10.1016/j.ijrobp.2023.12.046. [Google Scholar] [PubMed] [CrossRef]
89. Lieberman NAP, DeGolier K, Kovar HM, Davis A, Hoglund V, Stevens J, et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: implications for development of immunotherapy. Neuro Oncol. 2019;21(1):83–94. doi:10.1093/neuonc/noy145. [Google Scholar] [PubMed] [CrossRef]
90. Pesce S, Greppi M, Ferretti E, Obino V, Carlomagno S, Rutigliani M, et al. miRNAs in NK cell-based immune responses and cancer immunotherapy. Front Cell Dev Biol. 2020;8:119. doi:10.3389/fcell.2020.00119. [Google Scholar] [PubMed] [CrossRef]
91. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol. 2019;16(5):430–41. doi:10.1038/s41423-019-0206-4. [Google Scholar] [PubMed] [CrossRef]
92. Kennedy PR, Arvindam US, Phung SK, Ettestad B, Feng X, Li Y, et al. Metabolic programs drive function of therapeutic NK cells in hypoxic tumor environments. Sci Adv. 2024;10(44):1–23. doi:10.1126/sciadv.adn1849. [Google Scholar] [PubMed] [CrossRef]
93. Kaur K, Jewett A. Combination therapy of supercharged NK cells and ONC201 or ONC206 to target aggressive K27M brain tumor. Crit Rev Immunol. 2025;45(3). doi:10.1615/CritRevImmunol.2025058345. [Google Scholar] [PubMed] [CrossRef]
94. Jewett A, Kaur K, Gharamanians N. Supercharged NK cells as a promising therapeutic strategy to target and eliminate aggressive DIPG tumors in pediatric patients. Crit Rev Immunol. 2025;45(4):13–6. doi:10.1615/CritRevImmunol.2025059050. [Google Scholar] [PubMed] [CrossRef]
95. Poorva P, Mast J, Cao B, Shah MV, Pollok KE, Shen J. Killing the killers: natural killer cell therapy targeting glioma stem cells in high-grade glioma. Mol Ther. 2025;33(6):2462–78. doi:10.1016/j.ymthe.2025.02.043. [Google Scholar] [PubMed] [CrossRef]
96. Li H, Song W, Li Z, Zhang M. Preclinical and clinical studies of CAR-NK-cell therapies for malignancies. Front Immunol. 2022;13:992232. doi:10.3389/fimmu.2022.992232. [Google Scholar] [PubMed] [CrossRef]
97. Page A, Chuvin N, Valladeau-Guilemond J, Depil S. Development of NK cell-based cancer immunotherapies through receptor engineering. Cell Mol Immunol. 2024;21(4):315–31. doi:10.1038/s41423-024-01145-x. [Google Scholar] [PubMed] [CrossRef]
98. Jørgensen LV, Christensen EB, Barnkob MB, Barington T. The clinical landscape of CAR NK cells. Exp Hematol Oncol. 2025;14(1):46. doi:10.1186/s40164-025-00633-8. [Google Scholar] [PubMed] [CrossRef]
99. Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: a promising cellular immunotherapy for cancer. EBioMedicine. 2020;59(3):102975. doi:10.1016/j.ebiom.2020.102975. [Google Scholar] [PubMed] [CrossRef]
100. Jünemann W, Bley I, Rekowski L, Klokow M, Herppich S, Müller I, et al. GD2-CAR NK-92 cell activity against neuroblastoma cells is insusceptible to TIGIT knockout. Cancer Immunol Immunother. 2025;74(6):191. doi:10.1007/s00262-025-04010-6. [Google Scholar] [PubMed] [CrossRef]
101. Zuo P, Li Y, He C, Wang T, Zheng X, Liu H, et al. Anti-tumor efficacy of anti-GD2 CAR NK-92 cells in diffuse intrinsic pontine gliomas. Front Immunol. 2023;14:1145706. doi:10.3389/fimmu.2023.1145706. [Google Scholar] [PubMed] [CrossRef]
102. Mitwasi N, Feldmann A, Arndt C, Koristka S, Berndt N, Jureczek J, et al. UniCAR-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci Rep. 2020;10(1):2141. doi:10.1038/s41598-020-59082-4. [Google Scholar] [PubMed] [CrossRef]
103. Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, et al. Chimeric antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol. 2017;8:533. doi:10.3389/fimmu.2017.00533. [Google Scholar] [PubMed] [CrossRef]
104. Zhang M, Lam KP, Xu S. Natural killer cell engagers (NKCEsa new frontier in cancer immunotherapy. Front Immunol. 2023;14:1207276. doi:10.3389/fimmu.2023.1207276. [Google Scholar] [PubMed] [CrossRef]
105. Nikkhoi SK, Li G, Hatefi A. Natural killer cell engagers for cancer immunotherapy. Front Oncol. 2025;14:1483884. doi:10.3389/fonc.2024.1483884. [Google Scholar] [PubMed] [CrossRef]
106. Si C, Gao J, Ma X. Natural killer cell-derived exosome-based cancer therapy: from biological roles to clinical significance and implications. Mol Cancer. 2024;23(1):134. doi:10.1186/s12943-024-02045-4. [Google Scholar] [PubMed] [CrossRef]
107. Zhang A, Yin X, Ma J. NK-derived exosomes in anti-tumor strategies. Med Oncol. 2025;42(9):418. doi:10.1007/s12032-025-02965-1. [Google Scholar] [PubMed] [CrossRef]
108. Afolabi LO, Bi J, Li X, Adeshakin AO, Adeshakin FO, Wu H, et al. Synergistic tumor cytolysis by NK cells in combination with a pan-HDAC inhibitor, panobinostat. Front Immunol. 2021;12:701671. doi:10.3389/fimmu.2021.701671. [Google Scholar] [PubMed] [CrossRef]
109. Cheng B, Pan W, Xiao Y, Ding Z, Zhou Y, Fei X, et al. HDAC-targeting epigenetic modulators for cancer immunotherapy. Eur J Med Chem. 2024;265:116129. doi:10.1016/j.ejmech.2024.116129. [Google Scholar] [PubMed] [CrossRef]
110. Galat Y, Du Y, Perepitchka M, Li XN, Balyasnikova IV, Tse WT, et al. In vitro vascular differentiation system efficiently produces natural killer cells for cancer immunotherapies. Oncoimmunology. 2023;12(1):2240670. doi:10.1080/2162402X.2023.2240670. [Google Scholar] [PubMed] [CrossRef]
111. Zhong Y, Liu J. Emerging roles of CAR-NK cell therapies in tumor immunotherapy: current status and future directions. Cell Death Discov. 2024;10(1):318. doi:10.1038/s41420-024-02077-1. [Google Scholar] [PubMed] [CrossRef]
112. Zhang Y, Hu R, Xie X, Li Y. Expanding the frontier of CAR therapy: comparative insights into CAR-T, CAR-NK, CAR-M, and CAR-DC approaches. Ann Hematol. 2025;104(9):4305–17. doi:10.1007/s00277-025-06538-0. [Google Scholar] [PubMed] [CrossRef]
113. Fan Z, Han D, Fan X, Zhao L. Ovarian cancer treatment and natural killer cell-based immunotherapy. Front Immunol. 2023;14:1308143. doi:10.3389/fimmu.2023.1308143. [Google Scholar] [PubMed] [CrossRef]
114. Yang Y, Lundqvist A. Immunomodulatory effects of IL-2 and IL-15; implications for cancer immunotherapy. Cancers. 2020;12(12):3586. doi:10.3390/cancers12123586. [Google Scholar] [PubMed] [CrossRef]
115. Cao Y, Wang X, Jin T, Tian Y, Dai C, Widarma C, et al. Immune checkpoint molecules in natural killer cells as potential targets for cancer immunotherapy. Signal Transduct Target Ther. 2020;5(1):250. doi:10.1038/s41392-020-00348-8. [Google Scholar] [PubMed] [CrossRef]
116. Khan M, Arooj S, Wang H. NK cell-based immune checkpoint inhibition. Front Immunol. 2020;11:167. doi:10.3389/fimmu.2020.00167. [Google Scholar] [PubMed] [CrossRef]
117. Buckle I, Guillerey C. Inhibitory receptors and immune checkpoints regulating natural killer cell responses to cancer. Cancers. 2021;13(17):4263. doi:10.3390/cancers13174263. [Google Scholar] [PubMed] [CrossRef]
118. Kuznetsova AV, Glukhova XA, Beletsky IP, Ivanov AA. NK cell activity in the tumor microenvironment. Front Cell Dev Biol. 2025;13:1609479. doi:10.3389/fcell.2025.1609479. [Google Scholar] [PubMed] [CrossRef]
119. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. New Engl J Med. 2020;382(6):545–53. doi:10.1056/NEJMoa1910607. [Google Scholar] [PubMed] [CrossRef]
120. Becker PSA, Suck G, Nowakowska P, Ullrich E, Seifried E, Bader P, et al. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol Immunother. 2016;65(4):477–84. doi:10.1007/s00262-016-1792-y. [Google Scholar] [PubMed] [CrossRef]
121. Duygu B, Olieslagers TI, Groeneweg M, Voorter CEM, Wieten L. HLA class I molecules as immune checkpoints for NK cell alloreactivity and anti-viral immunity in kidney transplantation. Front Immunol. 2021;12:680480. doi:10.3389/fimmu.2021.680480. [Google Scholar] [PubMed] [CrossRef]
122. Ruggeri L, Vago L, Eikema DJ, de Wreede LC, Ciceri F, Diaz MA, et al. Natural killer cell alloreactivity in HLA-haploidentical hematopoietic transplantation: a study on behalf of the CTIWP of the EBMT. Bone Marrow Transplant. 2021;56(8):1900–7. doi:10.1038/s41409-021-01259-0. [Google Scholar] [PubMed] [CrossRef]
123. Zhou J, Zhang S, Guo C. Crosstalk between macrophages and natural killer cells in the tumor microenvironment. Int Immunopharmacol. 2021;101(Pt B):108374. doi:10.1016/j.intimp.2021.108374. [Google Scholar] [PubMed] [CrossRef]
124. Shaffer BC, Hsu KC. Selection of allogeneic hematopoietic cell transplant donors to optimize natural killer cell alloreactivity. Semin Hematol. 2020;57(4):167–74. doi:10.1053/j.seminhematol.2020.10.005. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools