
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

HBx Protein in Hepatitis B Virus-Related Hepatocellular Carcinoma: Pathogenic Mechanisms and Emerging Interventions
1 Department of Research, Taipei Tzu Chi Hospital, The Buddhist Tzu Chi Medical Foundation, New Taipei City, 231, Taiwan
2 Department of Internal Medicine, Cathay General Hospital, Taipei, 106, Taiwan
3 Taipei Fuhsing Private School, Taipei, 106, Taiwan
4 Department of Dentistry, Taipei Tzu Chi Hospital, The Buddhist Tzu Chi Medical Foundation, New Taipei City, 231, Taiwan
5 Taipei American School, Taipei, 111, Taiwan
6 Division of Nephrology, Department of Internal Medicine, Sijhih Cathay General Hospital, New Taipei City, 221, Taiwan
7 Department of Medical Research, Cathay General Hospital, Taipei, 106, Taiwan
8 Institute of Oral Medicine and Materials, College of Medicine, Tzu Chi University, Hualien, 970, Taiwan
* Corresponding Authors: Po-Chih Hsu. Email: ; Chan-Yen Kuo. Email:
# These authors contributed equally to this work
BIOCELL 2026, 50(3), 1 https://doi.org/10.32604/biocell.2025.073698
Received 23 September 2025; Accepted 21 November 2025; Issue published 23 March 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Hepatocellular carcinoma (HCC) is a leading cause of cancer-related death worldwide, most commonly driven by chronic hepatitis B virus (HBV) infection. The HBV X protein (HBx) plays a central role in hepatocarcinogenesis by regulating transcription, signal transduction, epigenetic modification, and interactions with noncoding RNAs. This review summarizes current advances in HBx-mediated signaling pathways and mutation-specific functions, highlighting its potential as a prognostic biomarker and therapeutic target, and providing insights for future strategies in HCC treatment and HBV eradication. Activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), cAMP response element binding protein/activating transcription factor (CREB/ATF), and phosphatidylinositol 3′-kinase/AKT serine-threonine protein kinase family (PI3K/Akt) pathways by HBx promotes tumor proliferation, epithelial–mesenchymal transition, and immune evasion. Mutation- and truncation-specific variants of HBx, such as C1485T, C1653T, and K130M/V131I, further enhance oxidative stress, inflammatory signaling, and chemoresistance, contributing to poor prognosis. Emerging preclinical evidence indicates that natural compounds, including asiatic acid, sphondin, and rapamycin, can suppress HBx stability and transcriptional activity, offering novel antiviral and antitumor strategies. Understanding HBx-driven molecular mechanisms and mutation-specific effects may guide the development of precise diagnostic, prognostic, and therapeutic approaches for HBV-related HCC.Graphic Abstract
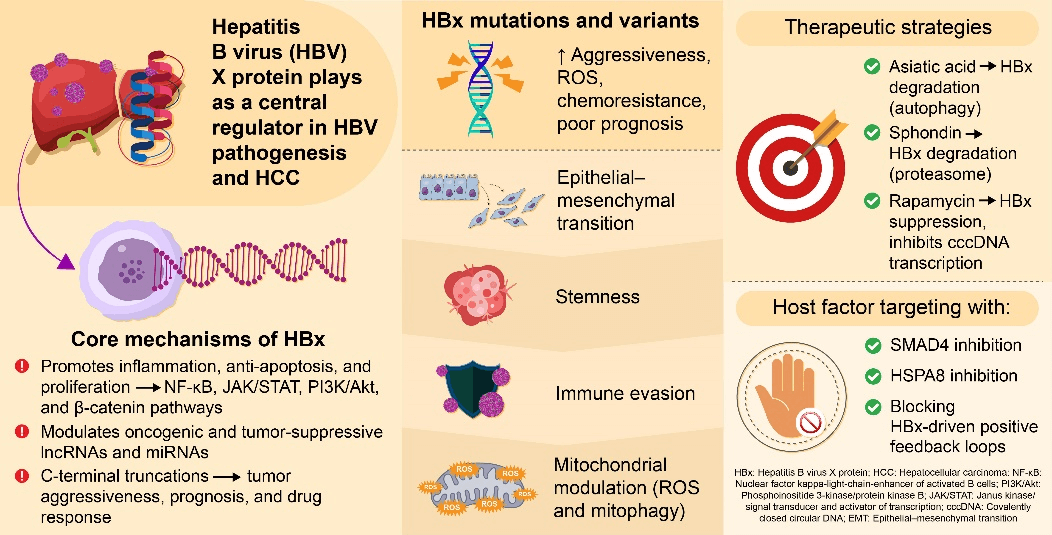
Keywords
The most common primary liver malignancy, hepatocellular carcinoma (HCC), is one of the leading global causes of death related to cancer [1]. The risk of HCC development notably increases during chronic infection with the hepatitis B virus (HBV) [2]. The HBV X protein (HBx) has garnered attention for its multifunctional roles in hepatocarcinogenesis. HBx comprises 154 amino acids and exhibits no intrinsic enzymatic activity; instead, it exerts its effects through interactions with host cellular machinery.
This narrative review was developed through a comprehensive literature search conducted in PubMed, Scopus, and Web of Science databases, covering studies published from January 2000 to September 2025. The search combined the keywords HBx, hepatitis B virus X protein, hepatocellular carcinoma, and molecular mechanism. Original research articles, reviews, and meta-analyses focusing on the pathogenic mechanisms, mutations, and therapeutic targeting of HBx in HBV-related hepatocellular carcinoma were included. Non-English papers and studies unrelated to HBx-mediated pathways were excluded [3].
Herein, we provide an overview to encompass the functional roles of this elusive protein and discuss potential therapeutic approaches for its targeted inhibition.
2 Structure and Localization of HBx
The X gene of HBV encodes HBx, and it is expressed during both acute and chronic phases of infection, significantly contributing to HBV pathogenesis and the development of HCC [4]. HBx comprises 154 amino acids and has a molecular weight of approximately 17 kDa. This multifunctional regulatory protein is known for its ability to localize to numerous cellular compartments (e.g., cytoplasm, nucleus, and mitochondria) where it modulates various cellular activities that include transcription, signal transduction, apoptosis, and DNA repair [5]. Structurally, HBx does not possess a classical DNA-binding domain, which limits its direct interaction with genomic DNA. Instead, its function is largely mediated through protein-protein interactions with various host factors [6]. The 1–50-residue-long N-terminal domain is relatively unstructured and flexible, allowing it to dynamically interact with signaling molecules and participate in the transcriptional activation of various genes. This domain contributes to the transactivation potential of HBx by engaging with transcription factors and coactivators, such as CREB-binding protein (CBP) and p300 [3,7]. In contrast, the C-terminal domain (residues 51–154) is more structurally conserved and plays a central role in mediating interactions of HBx with cellular and viral components. This region contains multiple motifs essential for oncogenic activity, including mitochondrial localization sequences and domains required for the modulation of various signaling pathways, including the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway [3,8,9]. Notably, the C-terminal domain facilitates the subcellular trafficking of HBx, enabling its translocation between the cytoplasm and nucleus in response to cellular cues and stress signals [10].
HBx localization to the mitochondria has been particularly linked to its role in regulating apoptosis and reactive oxygen species (ROS) production. HBx interacts with mitochondrial proteins, such as voltage-dependent anion channel 3 (VDAC3), influencing the mitochondrial membrane potential and promoting oxidative stress, which can contribute to genomic instability and tumorigenesis [11,12]. Additionally, nuclear localization of HBx enables it to interact with components of transcriptional machinery (e.g., transcription factor II B (TFIIB), transcription factor II H (TFIIH), CREB-binding protein (CBP)/p300, and other transcription factors) to modulate host and viral transcription, thereby altering gene expression and DNA repair processes. In contrast, cytoplasmic HBx engages and activates key signaling cascades, such as the Src kinase, Janus kinase/signal transducer and activator of transcription (JAK/STAT), and phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) pathways, to promote oncogenic signaling [13–15].
Together, the structural modularity and dynamic subcellular distribution of HBx underpin its ability to manipulate host cell function and drive oncogenic transformation during HBV infection. Mutations and truncations, particularly in the C-terminal region, are associated with enhanced tumorigenic potential and chemoresistance in HCC, further highlighting the importance of HBx structure-function relationships in the progression of HBV-related liver disease (Fig. 1).
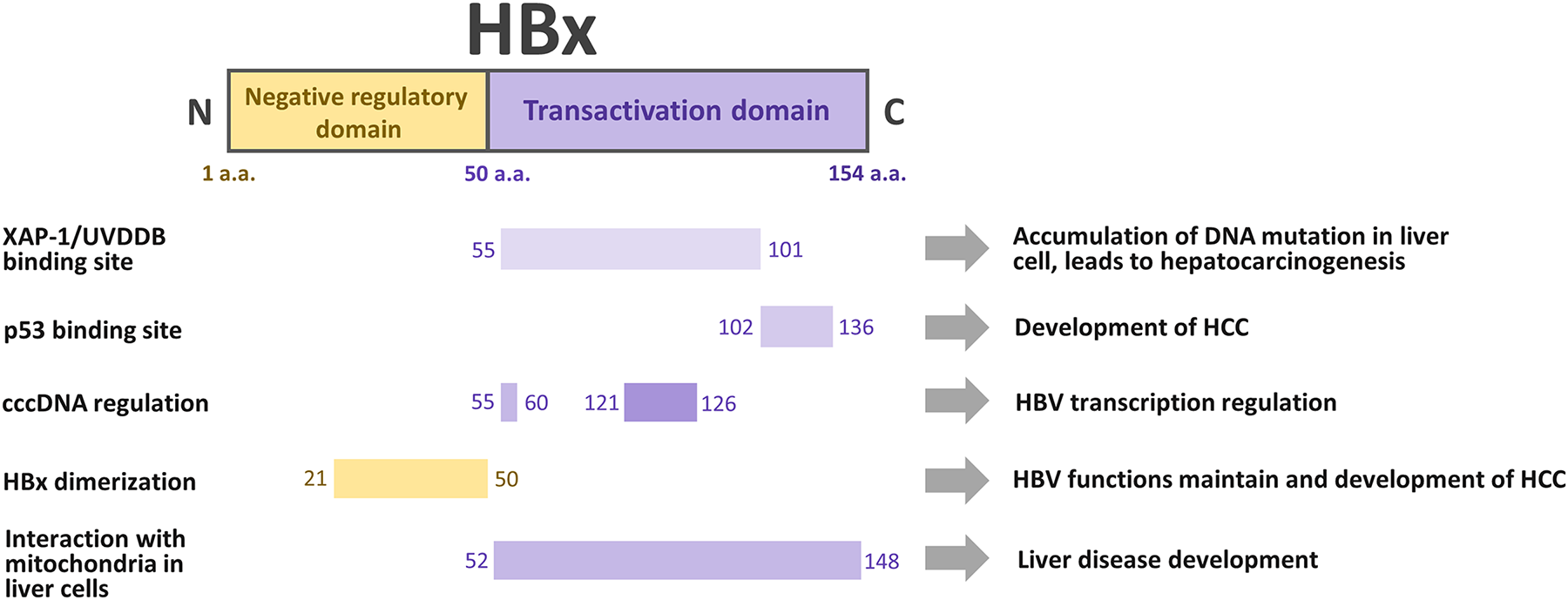
Figure 1: Interactions of hepatitis B virus X protein (HBx) domains with cellular proteins in host liver cells. The 154-amino-acid (a.a.) long HBx protein is divided into two major regions, the N-terminal negative regulatory (1–50 a.a.) [3,7] and C-terminal transactivation (50–154 a.a.) domains [3,8,9]. Key sites include XAP-1/UV-DDB (55–101 a.a.), p53 (102–136 a.a.), cccDNA regulation (55–60, 121–126 a.a.), dimerization (21–50 a.a.), and mitochondrial interaction (52–148 a.a.), which collectively drive hepatitis B virus (HBV) transcription, viral persistence, and hepatocellular carcinoma (HCC) development
3 Molecular Mechanisms of HBx in HCC
3.1 Transcriptional Dysregulation by HBx: A Key Driver of Hepatocarcinogenesis
The ability of HBx to modulate host gene expression at the transcriptional level is its most extensively studied feature [16]. Although HBx does not possess intrinsic DNA-binding activity, it exerts powerful transactivation effects through interactions with a variety of nuclear transcription factors, coactivators, and chromatin-modifying complexes [10,17]. This transcriptional dysregulation plays a pivotal role in HBV replication and HCC initiation and progression [18]. HBx enhances transcription by interacting with components of the basal transcription machinery, such as RNA polymerase II, TATA-binding protein, and transcription factor IIB [8]. These interactions facilitate recruitment of the transcriptional complex to target promoters, leading to the aberrant expression of key genes that affect cell proliferation and survival [19]. These interactions, in turn, activate or repress multiple signaling pathways that contribute to hepatocarcinogenesis.
3.2 Multifaceted Activation of NF-κB Signaling by HBx in Hepatocarcinogenesis: Roles of TBK1, Prion Protein Condensates, and SHP2
HBx activates the NF-κB signaling pathway, a key regulator of inflammation, immune response, and cell survival [9,20]. This activation is partly mediated by the proteasomal degradation of IκB, an NF-κB inhibitor, allowing the p65 subunit to translocate into the nucleus. Therein, NF-κB promotes the transcription of target genes involved in anti-apoptosis (including B-cell lymphoma-extra large [Bcl-xL], inflammation (including interleukin [IL]-6), TNF-α, and cell cycle regulation [21]. A previous study revealed that HBx enhances NF-κB signaling by transcriptionally upregulating TANK-binding kinase 1 (TBK1), which in turn promotes NF-κB p65 phosphorylation at serine 536. The study findings suggest that TBK1 may contribute to HBx-mediated HCC development through sustained NF-κB activation [22]. Guo et al. reported that HBx contributes to NF-κB activation and nuclear translocation in HCC, implicating HBx in abnormal NF-κB signaling during hepatocarcinogenesis [23]. HBx enhances cellular prion protein (PrPC) expression via cAMP response element-binding protein 1. PrPC forms condensates that recruit components of the NF-κB pathway-TRAF2/6, TAB2/3, and TAK1-activating NF-κB signaling and promoting tumor growth. The α3 helix and disulfide-bond formation in PrPC are crucial for liquid-liquid phase separation and NF-κB activation. This, in turn, elevates IL-8 expression, further contributing to tumor progression [24]. HBx and NF-κB upregulate Src homology region 2-containing protein tyrosine phosphatase 2 (SHP2) by promoting NF-κB binding to the promoter of Shp2. SHP2 facilitates extracellular signal-regulated kinase (ERK) activation via complex formation with EGFR and Gab1. In vitro, SHP2-ERK activation negatively correlates with signal transducer and activator of transcription 3 (STAT3) phosphorylation. In HBV-related HCC tissues, SHP2 expression is reduced compared with that in adjacent non-tumorous liver, with the highest levels observed in fibrotic background tissue. SHP2 expression progressively increases during liver disease progression but is lost in dysplastic and cancerous tissues. These findings suggest that SHP2 depletion disrupts NF-κB-STAT3 crosstalk, promoting HCC development [25]. Together, these findings underscore the complex regulatory network orchestrated by HBx-NF-κB signaling and highlight potential molecular targets for therapeutic intervention in HBV-associated HCC [26]. Table 1 summarizes the mechanisms by which HBx activates the NF-κB signaling pathway, highlighting the molecular intermediates involved, downstream gene expression changes, and their functional contributions to hepatocarcinogenesis.

To enhance clarity and clinical orientation, we summarized the principal HBx-mediated NF-κB signaling mechanisms in Table 1. This structured overview highlights only the most functionally and clinically relevant intermediates such as TBK1, cellular prion protein (PrPC) condensates, and SHP2 that contribute to hepatocellular inflammation, fibrosis, and tumor progression. By focusing on these major regulatory nodes, we aimed to facilitate readers’ understanding of how HBx-induced NF-κB activation directly links to hepatocarcinogenesis and therapeutic opportunities.
3.3 Multifaceted Oncogenic Roles of HBx in HCC via the CREB/ATF and NF-κB/S100A9 Pathways
CREB and the activating transcription factor (ATF) family are pivotal regulators of gene expression involved in cell growth, survival, metabolism, and inflammation [27,28]. HBx activates the CREB/ATF pathway, contributing to HCC development [29]. Cougot et al. demonstrated that HBx physically binds CBP/p300 in vitro and in vivo, and it cooperatively enhances CREB-mediated transcription, particularly when CREB is phosphorylated by protein kinase A, which may promote the initiation of tumorigenesis [30]. Duan et al. reported that HBx promotes the nuclear translocation of NF-κB, which in turn enhances S100A9 transcription. Functional assays confirmed that S100A9 contributes to HCC cell growth and metastasis induced by HBx. Clinically, S100A9 levels in serum were associated with extrahepatic metastasis, TNM stage, and HBV DNA load, and they showed diagnostic potential for identifying metastatic disease [31]. A previous study found that HBx initially activates transcription of the tumor suppressor gene, Ras association domain family 1 isoform A (RASSF1A), via SP1 but contributes to its silencing through promoter methylation, ultimately facilitating HCC progression. [32]. Collectively, these findings highlight the multifaceted role that HBx plays in HCC development. HBx promotes HCC development through multiple transcriptional pathways, notably by activating the CREB/ATF axis via CBP/p300 interaction and inducing S100A9 expression through NF-κB activation. These mechanisms contribute to tumor growth and metastasis, highlighting the potential of using S100A9 as a relevant diagnostic biomarker for HBV-related HCC progression and metastasis. Table 2 summarizes the mechanisms by which HBx triggers the NF-κB signaling pathway, highlighting the molecular intermediates involved, downstream gene expression changes, and their functional contributions to hepatocarcinogenesis.

Given the mechanistic diversity of non-coding RNA (ncRNA) regulation by HBx, we streamlined this section to emphasize axes with clear translational implications. The summarized molecular networks presented in Tables 2–4 illustrate HBx-mediated mechanisms involved in hepatocellular carcinoma progression, prognosis, and drug responsiveness, including CREB/ATF activation, tumor microenvironment modulation, and the regulation of microRNAs and lncRNAs most strongly associated with these cancer-related outcomes. This table-based presentation allows readers to appreciate key oncogenic pathways without excessive textual detail while retaining mechanistic completeness for reference.
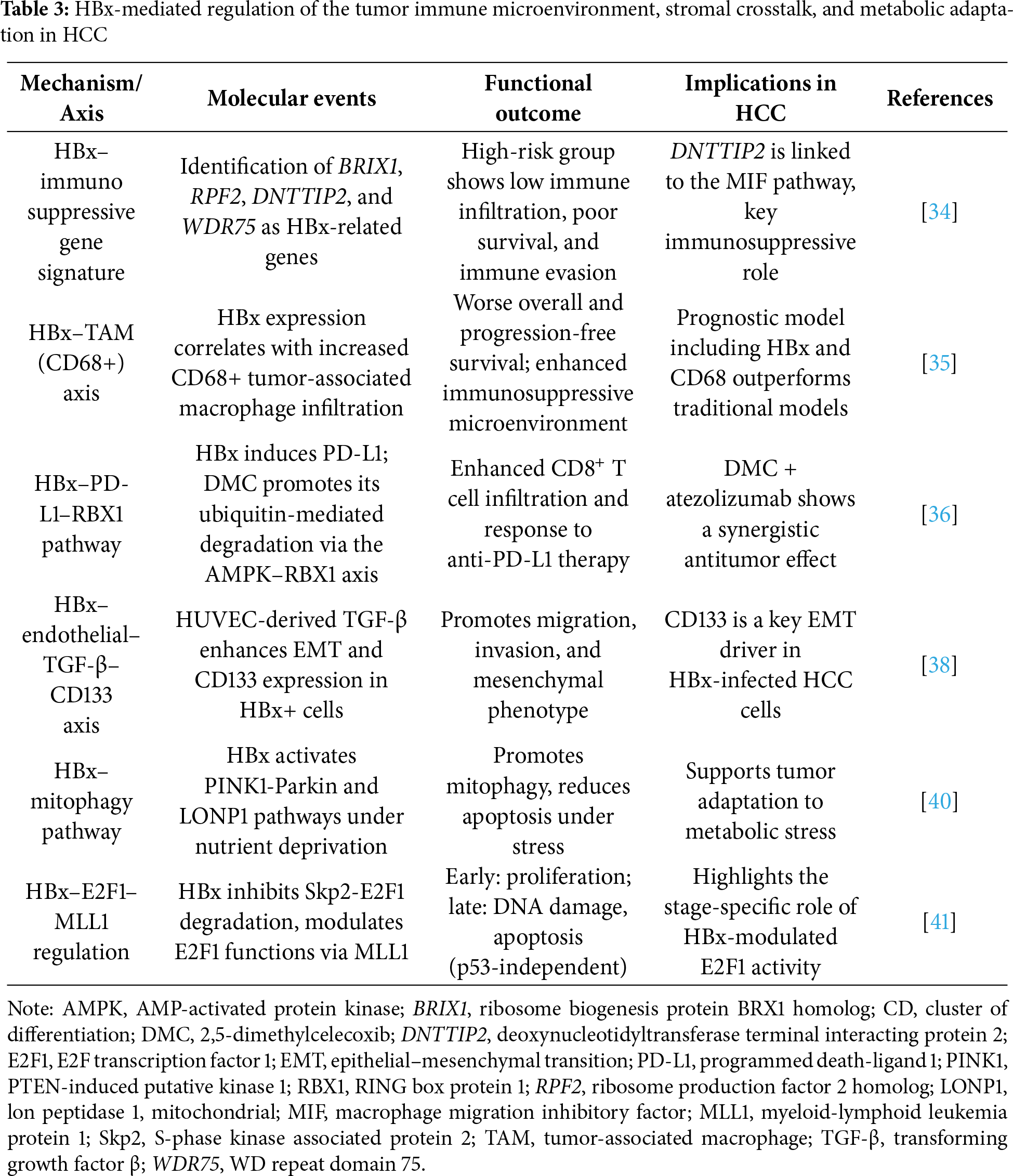

3.4 Multifaceted Role of HBx in Modulating the Tumor Immune Microenvironment and Progression of HBV-Related HCC
Understanding the complex interplay between the tumor immune microenvironment, metabolism, and gut microbiota is essential to overcoming immune evasion and resistance in HCC [33]; however, the role HBx plays in regulating the tumor immune microenvironment remains unclear. Zhong et al. investigated how HBx affects the tumor microenvironment (TME) and tumor immunity in HCC by identifying HBx-related genes and constructing a prognostic model. Transcriptomic analysis of HBx-expressing HepG2 cells, along with The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) data, revealed seven HBx-associated genes, with four (ribosome biogenesis protein BRX1 homolog [BRIX1], ribosome production factor 2 homolog [RPF2], deoxynucleotidyltransferase terminal interacting protein 2 [DNTTIP2], and WD repeat domain 75 [WDR75]) forming a prognostic risk score signature. Patients at high risk exhibited poorer survival, weaker immunotherapy responsiveness, reduced anti-tumor immune infiltration, and increased immune evasion. Among these, DNTTIP2 emerged as a key player linked to an immunosuppressive microenvironment through migration inhibitory factor signaling [34]. Increased CD68+ TAM infiltration in HCC tissues is closely associated with HBx expression. Elevated levels of either marker predicted worse overall and progression-free survival after hepatectomy, with the poorest outcomes observed in patients showing high expression of both. A prognostic model combining HBx, CD68, tumor size, and microvascular invasion demonstrated the highest predictive accuracy, outperforming conventional HCC classifications [35]. A previous study showed that HBV-related HCC exhibited lower CD8+ T cell levels and higher programmed death-ligand 1 (PD-L1) and CD163 expression levels compared with those in non-virus-related HCC. In mouse models, 2,5-dimethylcelecoxib (DMC) treatment elevated CD8+ T cell infiltration and lowered PD-L1 and CD163 levels in HBx-positive tumors. DMC combined with atezolizumab enhanced anti-tumor effects and more effectively inhibited the PD-1/PD-L1 pathway. Mechanistically, DMC promotes the ubiquitin-driven degradation of HBx-induced PD-L1 via AMP-activated protein kinase (AMPK) pathway activation, primarily through the E3 ligase, RING box protein 1 (RBX1) [36].
The TME comprises hepatic stellate cells (HSCs), macrophages, and endothelial cells, and it actively promotes tumor development while tumor cells further modulate the stroma to support their growth [37]. Rawal et al. demonstrated that endothelial cell-derived factors (particularly transforming growth factor β [TGF-β]) promote aggressive behavior in hepatoma cells infected with HBx. Co-culture with human umbilical vein endothelial cell (HUVEC)-conditioned media enhanced migration, invasion, and mesenchymal gene expression, particularly increasing CD133 levels. TGF-β stimulation replicated these effects, whereas CD133 knockdown reduced mesenchymal traits, highlighting CD133 as a key driver of the epithelial–mesenchymal transition (EMT) in cells infected with HBx. The findings suggest that endothelial-derived TGF-β contributes to HBV-related HCC progression by inducing the EMT through CD133 [38]. The formation of a tumor immune barrier (TIB) by secreted phosphoprotein 1 (SPP1) + macrophages and cancer-associated fibroblasts (CAFs) at the tumor boundary impairs immune checkpoint blockade efficacy by restricting immune cell infiltration. Hypoxia-induced SPP1 promotes macrophage CAF interactions that remodel the extracellular matrix and reinforce the TIB. Blocking SPP1 or deleting it in macrophages enhances anti-PD-1 therapy in mouse liver cancer by reducing CAFs and boosting the infiltration of cytotoxic T cells [39]. Another study revealed that HBx enhances mitophagy in nutrient-deprived HCC cells by upregulating the PINK1-Parkin pathway. HBx promotes Parkin recruitment to mitochondria and modulates the mitochondrial unfolded protein response through LONP1, boosting mitophagy and reducing apoptosis under starvation. These findings highlight a novel role of HBx in helping tumor cells adapt to metabolic stress, offering new insights into HCC progression [40]. Moreover, another study uncovered how HBx modulates E2F transcription factor 1 (E2F1) functions in HCC. HBx interferes with Skp2 binding, leading to E2F1 and MLL1 accumulation. In early tumor stages, E2F1 promotes proliferation, whereas in later stages it induces DNA damage and apoptosis independently of p53. These opposing roles are regulated by the dynamic promoter occupancy of MLL1 and its interaction with co-activators or repressors, revealing a mechanism for the stage-specific functions of E2F1 in tumor progression [41]. Collectively, these findings underscore HBx as a central driver of HCC progression through its regulation of stromal interactions, metabolic adaptation, immune evasion, and cell fate determination. Table 3 summarizes current findings on how HBx influences the TME and contributes to HCC progression.
3.5 Multifaceted Oncogenic Roles of HBx in HCC: Molecular Mechanisms and Therapeutic Implications
MicroRNAs (miRNAs) are emerging as crucial regulators of HCC pathogenesis that influence key cellular processes and cancer hallmarks [42]. A previous study revealed that HBx promotes tumor progression in HBV-related HCC by upregulating von Willebrand factor (vWF) expression, thereby enhancing proliferation, migration, and the EMT while also inhibiting apoptosis. Mechanistically, HBx increases vWF expression by upregulating achaete-scute family bHLH transcription factor 1 (ASCL1), a process mediated by the suppression of miR-3150b-3p and activation of the long noncoding RNA (lncRNA), ST8SIA6-AS1, which sponges miR-3150b-3p. These findings uncovered the novel HBx/ST8SIA6-AS1/miR-3150b-3p/ASCL1/vWF axis that contributes to the malignancy of HBV-related HCC [43]. Zhou et al. reported that the overexpression of spastic paraplegia 21 (SPG21) in HCC is associated with a poor prognosis. HBx suppresses miR-128-3p, leading to SPG21 upregulation, which enhances the calcium influx mediated by transient receptor potential cation channel subfamily M member 7 (TRPM7) and activates the c-Jun N-terminal kinase (JNK) pathway. This signaling cascade promotes tumor growth, invasion, and resistance to doxorubicin-induced apoptosis [44].
Furthermore, another study demonstrated that HBx alters microRNA expression in HCC cells. HBx upregulates oncogenic miR-21 and miR-222 while significantly downregulating tumor-suppressive miR-22, miR-122, and miR-132. These changes suggest that HBx promotes HCC progression by modulating key miRNAs, and the expression patterns of these miRNAs may serve as prognostic markers in HBV-related liver disease [45]. Wu et al. revealed that HBx promotes HCC metastasis by epigenetically repressing the tumor-suppressive microRNA, lethal-7c microRNA (let-7c), via upregulation of enhancer of zeste homolog 2 (EZH2). This repression increases high-mobility group AT-hook 2 (HMGA2) expression, enhancing cancer cell migration. The findings suggest that the HBx/EZH2/let-7c/HMGA2 axis plays a crucial role in HCC progression caused by HBV and that let-7c and HMGA2 may serve as valuable diagnostic markers and therapeutic targets [46]. HBx also promotes the progression of HCC by upregulating exosomal miR-155, which in turn inhibits the tumor suppressor, phosphatase and tensin homolog (PTEN). Suppression of PTEN activates the PI3K/Akt pathway, enhancing tumor cell proliferation, invasion, and survival. A negative correlation between PTEN and miR-155 and a positive correlation between HBx and miR-155 were observed in HBV-positive HCC tissues [47]. Downregulated expression of miR-361 was found in HBV-related HCC tissues, HBx-expressing cells, and HBx-transgenic mice. The miRNA, miR-361, directly targets and suppresses the p65 subunit of NF-κB, the expression of which is upregulated by HBx. Restoration of miR-361 reduces p65 levels and inhibits HSC growth and migration, whereas p65 overexpression reverses these effects [48].
Song et al. demonstrated that in HCC, HBx upregulates the lncRNA, transcriptional regulatory RNA 1 (TRERNA1), which promotes tumor cell proliferation and sorafenib resistance. Mechanistically, TRERNA1 functions as a competing endogenous RNA, sponging miR-22-3p to upregulate neuroblastoma RAS viral oncogene homolog (NRAS) and activate the RAS/Raf/MEK/ERK signaling pathway [49]. HBx also plays a key role in promoting abnormal α-fetoprotein (AFP) expression in HBV-related HCC by downregulating miR-1236 and miR-329, which normally suppress AFP translation. Elevated AFP levels enhance HCC cell proliferation and reduce sensitivity to chemotherapy [50]. Liu et al. speculated that HCC up-regulated long non-coding RNA (HULC), an lncRNA highly expressed in HCC, promotes HBV replication by enhancing the stability of covalently closed circular DNA (cccDNA) through the downregulation of APOBEC3B. Mechanistically, HULC upregulates miR-539 (which targets APOBEC3B), and this process is driven by HBx/STAT3-mediated activation of the promoter of miR-539. Functionally, HULC enhances hepatoma cell growth by facilitating HBV activity in vitro and in vivo. These findings highlight a novel HULC-HBx/STAT3-miR-539-APOBEC3B axis that contributes to HBV-linked HCC progression [51]. HBx enhances the properties of cancer stem cells (CSCs) and tumorigenicity in HCC by upregulating stemness markers and reprogramming proteins via the PI3K/Akt pathway. HBx also increases metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) expression and decreases that of miR-124, which directly targets MALAT1. Restoring miR-124 or silencing MALAT1 inhibits CSC traits and tumorigenesis induced by HBx. This suggests that the HBx/MALAT1/miR-124 axis regulates CSC properties through PI3K/Akt signaling in HBV-related HCC [52]. Overall, these findings highlight the central and multifaceted role HBx plays in driving HCC pathogenesis.
HBx contributes to HBV-related HCC progression by modulating a complex network of microRNAs, lncRNAs, transcription factors, and signaling pathways. It promotes tumor proliferation, invasion, the EMT, cancer stemness, and resistance to therapies, such as sorafenib and chemotherapy. Notably, HBx regulates key oncogenic axes, including the HBx/ST8SIA6-AS1/miR-3150b-3p/ASCL1/vWF, HBx/miR-128-3p/SPG21/TRPM7/JNK, HBx/EZH2/let-7c/HMGA2, HBx/MALAT1/miR-124/PI3K-Akt, and HBx/STAT3/miR-539/APOBEC3B axes, among others. Additionally, HBx influences tumor metabolism, immune evasion, and reactivation of fetal markers, such as AFP. These regulatory pathways not only offer mechanistic insights into HBx-mediated oncogenesis but also identify promising molecular targets and biomarkers that can facilitate the early detection, prognosis, and treatment of HBV-related HCC. Table 4 summarizes the key microRNAs, lncRNAs, and associated signaling pathways regulated by HBx in HBV-related HCC.
3.6 Targeting HBx: Emerging Therapeutic Strategies for Chronic Hepatitis B and HBV-Related HCC
Asiatic acid, a compound derived from Centella asiatica, is a novel anti-HBV agent that targets HBx. Asiatic acid promotes the degradation of HBx via autophagy, reduces its binding to cccDNA, induces repressive chromatin modifications, and suppresses the transcription of cccDNA. In both HBV-infected cells (in vitro) and a chronic infection mouse model (in vivo), asiatic acid significantly decreased HBV RNA, DNA, and HBsAg levels, demonstrating its potential as a promising candidate for HBx-targeted chronic hepatitis B therapy [53]. Ren et al. identified sphondin, a natural compound in traditional Chinese medicine, as a novel antiviral agent against HBV in vitro experiments. Sphondin selectively binds to HBx at Arg72, promoting its proteasome-mediated degradation. This impedes HBx recruitment to cccDNA, thereby inhibiting the transcription of cccDNA and HBsAg expression without affecting cccDNA levels. The antiviral effect is lost when HBx is absent or mutated [54]. Rapamycin has been reported to effectively inhibit HBx expression, and it exhibits notable anti-HBV activity, particularly by suppressing cccDNA transcription through the ubiquitin–proteasome system-mediated degradation of HBx in vitro and in vivo. These findings support rapamycin as a promising therapeutic candidate that could be used to develop a functional cure for chronic hepatitis B (CHB). However, it is important to note that rapamycin has a limited impact on HBV gene expression originating from integrated viral DNA, as this transcription is independent of cccDNA [55]. Similar results revealed high expression of SMAD family member 4 (SMAD4) in HBV-positive HCC, which is associated with poor prognosis. SMAD4 enhances HCC cell proliferation, and its effects are maintained even when HBx is knocked down and SMAD4 reintroduced. Mechanistically, HBx upregulates SMAD4 transcription via TFII-I and stabilizes it by inhibiting its ubiquitination through binding at the MH2 domain. Additionally, SMAD4 promotes HBx expression, forming a positive feedback loop [56]. HBx upregulates the expression of heat shock protein family A member 8 (HSPA8) via activation of the transcription factor, heat shock factor protein 1 (HSF1), in HCC. HSPA8 enhances HBV replication by recruiting HBx to the cccDNA minichromosome, forming a positive feedback loop, and concurrently suppresses ferroptosis by upregulating SLC7A11/GPX4, reducing ROS and Fe2+ accumulation. Inhibition of HSPA8 limits tumor growth and increases erastin sensitivity [57].
In summary, targeting HBx has emerged as a promising strategy for treating CHB and HBV-related HCC. Several natural compounds, including asiatic acid, sphondin, and rapamycin, have demonstrated the ability to inhibit HBx through distinct mechanisms, including autophagy or proteasome-mediated degradation, which lead to the reduced transcription of cccDNA and suppression of viral replication. In parallel, studies have shown that HBx interacts with host factors, such as SMAD4 and HSPA8, forming oncogenic positive feedback loops that influence HCC progression and resistance to cell death. Collectively, these findings highlight the potential benefits of disrupting HBx-associated pathways not only to control HBV replication but also to impede HBV-driven hepatocarcinogenesis, paving the way for novel, HBx-targeted interventions in CHB treatment. Table 5 summarizes the key natural compounds and molecular pathways that modulate HBx and their therapeutic implications. Additionally, Table 5 complements Figs. 1 and 2 by summarizing HBx-directed therapeutic interventions, including natural compounds (asiatic acid, sphondin, and rapamycin) and host-factor modulators (SMAD4 and HSPA8). It links each treatment to its mechanistic target and antiviral or anti-tumor effect, thereby providing a schematic bridge between HBx structure–function relationships (Fig. 1), mutation-specific pathogenic mechanisms (Fig. 2), and clinical translation through HBx-targeted therapy.

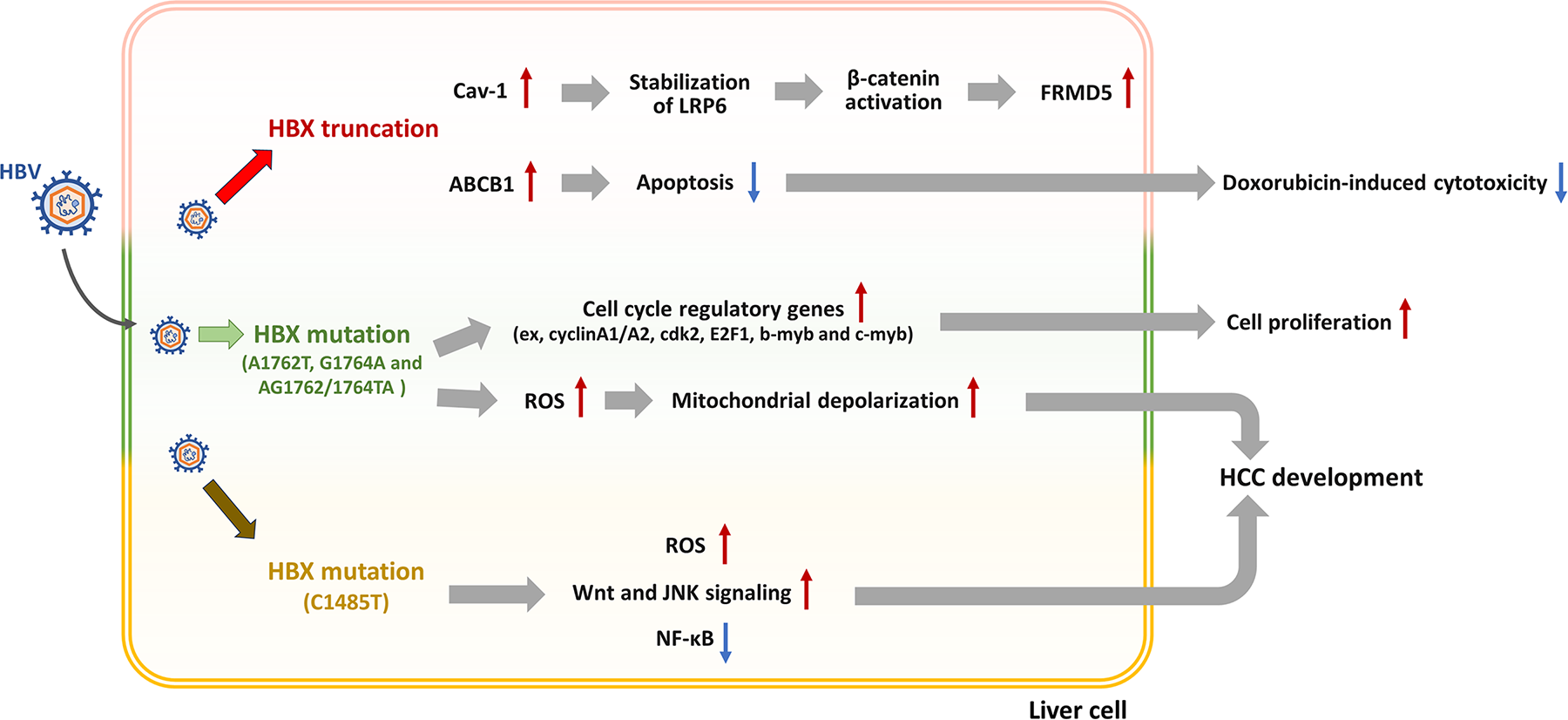
Figure 2: Truncation and mutations of the hepatitis B virus (HBV) X protein (HBx) contribute to the regulation of HBV-induced hepatocellular carcinoma (HCC) development. After HBV infects the liver cell, the truncated HBx enhances caveolin-1 (Cav-1)/β-catenin/FRMD5 signaling and ABCB1-mediated drug resistance, reducing doxorubicin cytotoxicity. HBx mutations dysregulate cell cycle regulators, elevate reactive oxygen species (ROS) levels, and induce mitochondrial depolarization. ROS further activates Src/c-Jun N-terminal kinase (JNK) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling. Collectively, these alterations promote apoptosis resistance, uncontrolled proliferation, and HCC development
3.7 Oncogenic Mechanisms and Therapeutic Implications of HBx Mutations in HBV-Associated HCC
A previous study evaluated how two HBx mutations (C1653T and T1753C) affect HCC progression. The C1653T mutation significantly enhanced HBx-induced proliferation, invasion, migration, and anti-apoptotic activity in HCC cells while also promoting tumor growth in vivo. Mechanistically, C1653T increased fibrosis and intracellular ROS levels and altered cytokine levels, including those of MCP-1 and IL-18. Drug sensitivity analysis revealed that C1653T responded well to apatinib, potentially due to increased VEGF expression. These findings suggest that this mutation contributes to HCC malignancy and may serve as a predictive biomarker for apatinib responsiveness [58]. Valosin-containing protein-interacting protein 1 (VCPIP1) was identified by Wu et al. as a novel deubiquitinating enzyme (DUB) that could stabilize HBx via a ubiquitin-independent mechanism. After VCPIP1 and HBx interact, the 26S proteasome subunit, PSMC3, is recruited to form a ternary complex that prevents HBx degradation by the 20S proteasome. This stabilization enhances the transcriptional activity of HBx and promotes the transcription of HBV cccDNA. These results reveal a new host-virus interaction pathway and suggest that targeting the VCPIP1-HBx-PSMC3 complex could be a potential strategy for developing DUB-based antiviral therapies [59]. A previous study investigated the role specific HBx mutations play in promoting HCC and aimed to identify novel therapeutic targets. Using Sleeping Beauty transposon-based mouse models, the researchers demonstrated that the M3 and Ct-HBx variants led to a higher incidence of HCC and enhanced cell proliferation and S phase progression compared with those of wild-type HBx. These mutants also induced greater expression of inflammatory cytokines and upregulated the gene expression levels of PAI1 (plasminogen activator inhibitor-1) and CDC20. Functional analysis revealed that when PAI1 is silences, the pro-proliferative effects of the mutants are attenuated, highlighting PAI1 as a potential mediator and candidate target in HBV-related HCC therapy [60]. Chiu et al. demonstrated that the K130M/V131I HBx variant promotes the progression of HCC by triggering the AKT/forkhead box O1 (FOXO1) axis and enhancing inflammatory signaling employing the Sleeping Beauty transposon system in Fah- and Trp53-deficient mice [61].
Collectively, these studies emphasize the importance of specific HBx mutations in driving HCC progression through distinct molecular mechanisms. Mutations such as C1653T and K130M/V131I enhance cell proliferation, invasion, and inflammatory signaling while also influencing drug responsiveness, as seen with the sensitivity of C1653T to apatinib. The identification of PAI1 as a key effector of M3 and Ct-HBx variants, and the discovery of VCPIP1 as a novel DUB stabilizing HBx, further underscore the complexity of HBx-mediated oncogenesis. These findings deepen our understanding of HBV-related hepatocarcinogenesis and suggest innovative avenues for targeted therapies, including mutation-specific prognostic markers, immune modulation, and DUB-based antiviral strategies. Table 6 summarizes the key mutations in HBx, along with recently identified molecular mechanisms that contribute to HCC progression.

3.8 Functional Characterization of C-Terminal Truncations and Point Mutations in HBx Reveals Distinct Oncogenic Pathways and Chemoresistance Mechanisms in HBV-Related HCC
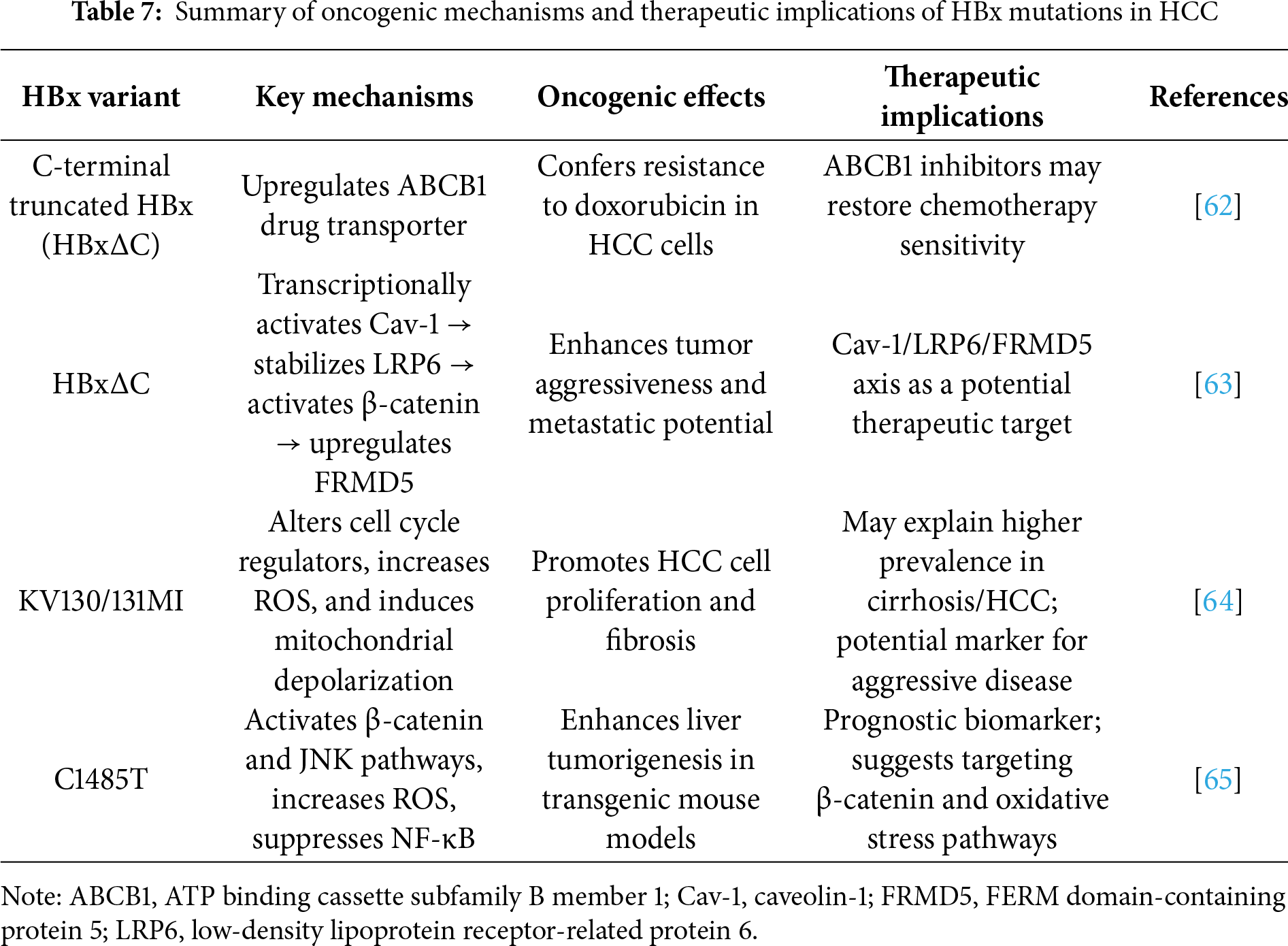
The C-terminal truncations and point mutations in HBx promote HBV-induced HCC development by regulating cell apoptosis and proliferation through different signaling pathways (Fig. 2). A previous study investigated how C-terminal truncated HBx variants mediate doxorubicin resistance in HCC. Truncated HBx-expressing cells exhibited decreased sensitivity to doxorubicin associated with upregulation of the drug efflux transporter, ABCB1. Functional inhibition or siRNA-mediated knockdown of ABCB1 restored doxorubicin-induced cytotoxicity, implicating ABCB1 as a key driver of resistance. These results suggest that co-administration of ABCB1 inhibitors with doxorubicin may enhance therapeutic efficacy in patients with HCC that exhibit C-terminal truncated HBx expression [62]. Mao et al. demonstrated that the C-terminal truncated form of HBx (HBxΔC) frequently identified in HCC, could enhance tumor aggressiveness by transcriptionally upregulating caveolin-1 (Cav-1). Cav-1 then promotes FERM domain-containing protein 5 (FRMD5) expression by stabilizing low-density lipoprotein receptor-related protein 6 (LRP6) and subsequently activating β-catenin signaling. Silencing Cav-1 or FRMD5 suppresses the oncogenic effects of HBxΔC, whereas overexpression of active β-catenin restores the tumorigenic potential. Correlated Cav-1, LRP6, and FRMD5 expression in clinical HCC samples underscores the significance of the HBxΔC/Cav-1/LRP6/FRMD5 axis as a candidate target for HBV-related HCC therapy [63].
The impact HBx mutants have on HCC progression has previously been investigated, with a focus on the KV130/131MI variant. In Huh7 cells, KV130/131MI markedly enhanced cell proliferation, altered cell cycle regulatory gene expression, elevated ROS levels, and induced mitochondrial depolarization. These results indicate that KV130/131MI may drive hepatocarcinogenesis and contribute to liver fibrosis by promoting unchecked proliferation and oxidative stress. The pronounced effects of this variant may account for its greater prevalence in patients affected by both cirrhosis and HCC when compared to the single A1762T or G1764A mutations [64]. A previous study identified the HBx gene mutations, C1485T and C1653T, as independent risk factors for HCC in patients with CHB. In transgenic mouse models, the C1485T-HBx variant significantly increased susceptibility to diethylnitrosamine-induced liver tumorigenesis compared with that of wild-type HBx. Mechanistically, this enhanced carcinogenic potential was linked to β-catenin and JNK signaling pathway activation, increased ROS production, and suppression of NF-κB activity. These findings demonstrate the direct contribution of the C1485T mutation in HBx to the development of HCC [65].
Taken together, these studies underscore the critical role of HBx mutations, particularly C-terminal truncations and point mutations, such as KV130/131MI, C1653T, and C1485T, in driving HCC progression through diverse oncogenic mechanisms. These include enhanced cell proliferation, resistance to chemotherapy via ABCB1 upregulation, β-catenin and JNK signaling activation, induction of oxidative stress, and suppression of tumor-suppressive pathways (e.g., NF-κB signaling pathway). The HBxΔC/Cav-1/LRP6/FRMD5 axis and Akt/FOXO1 pathway further highlight the molecular complexity by which HBx variants contribute to tumor aggressiveness and treatment resistance. These findings not only deepen our understanding of HBV-related hepatocarcinogenesis but also support the development of mutation-specific prognostic markers and targeted treatment strategies that could enhance therapeutic outcomes in patients with HCC. Table 7 summarizes key HBx variants implicated in HCC progression, highlighting their molecular mechanisms, oncogenic effects, and potential therapeutic implications. HBxΔC variants and specific point mutations, such as KV130/131MI and C1485T, contribute to diverse pathological features that include enhanced tumor proliferation, chemoresistance, the EMT, and immune evasion.
4 Conclusion and Future Perspective
HBx plays a multifaceted and central role in the pathogenesis of HCC, driving tumor initiation, progression, immune evasion, and therapy resistance. Through its interactions with host transcription factors, signaling pathways, epigenetic regulators, and noncoding RNAs, HBx orchestrates a complex regulatory network that promotes hepatocyte transformation and tumor aggressiveness. Specific HBx mutations, particularly C-terminal truncations and hotspot substitutions, such as C1485T, C1653T, and K130M/V131I, further enhance oncogenic signaling, alter tumor metabolism, modulate immune cell infiltration, and confer resistance to chemotherapy.
From a clinical perspective, emerging evidence indicates that HBx mutation profiling (including C1485T, C1653T, and K130M/V131I) may serve as an adjunct tool for diagnosis and prognostic evaluation in HBV-related HCC. Integration of HBx genotyping with conventional biomarkers such as serum HBV DNA load, alpha-fetoprotein (AFP) levels, and imaging features could enhance early detection and patient risk stratification. Furthermore, the overexpression of HBx and its truncated variants in tumor tissues correlates with aggressive clinicopathological characteristics, suggesting potential roles in guiding treatment intensity and surveillance intervals. From a therapeutic standpoint, the application of HBx-targeted compounds, including asiatic acid, sphondin, and rapamycin, holds promise as precision strategies complementing antiviral and immunomodulatory regimens. Continued validation of HBx-based molecular signatures in clinical cohorts will be essential for translating these mechanistic findings into personalized medicine approaches.
Moreover, the interplay between HBx and TME components, including immune cells, stromal factors, and gut microbiota, highlights the broader impact that the protein has beyond cell-autonomous mechanisms. Recent advances in targeting HBx through autophagy and proteasome-mediated degradation, and in identifying HBx-associated molecular axes, such as the HBx/CREB/PrPC/NF-κB, HBx/STAT3/miR-539, and HBxΔC/Cav-1/LRP6/FRMD5 axes, offer promising directions for therapeutic intervention.
Although remarkable progress has been made in elucidating the molecular mechanisms of HBx in HBV related hepatocarcinogenesis, several critical questions remain unresolved. Future research should clarify the temporal and spatial dynamics of HBx host interactions across disease stages and define the mutation specific oncogenic functions of variants such as K130M/V131I, C1485T, and C1653T. The interplay between HBx and the tumor immune microenvironment, including macrophage polarization, PD-L1 regulation, and cytokine signaling, requires deeper investigation to identify combinatorial immunotherapeutic targets. Moreover, HBx mediated epigenetic and noncoding RNA networks (e.g., HBx/EZH2/let 7c/HMGA2 and HBx/HULC/miR 539/APOBEC3B axes) warrant exploration as potential reversible mechanisms of tumor progression and drug resistance. The development of mutation tailored HBx inhibitors, vaccines, and RNA based therapeutics, validated through multi omics and translational models, represents a promising avenue toward personalized HBx directed therapy for HBV related HCC.
Collectively, this review underscores the importance of HBx as a diagnostic, prognostic, and therapeutic target in HBV-related HCC. Continued efforts to refine HBx-targeted strategies, delineate its signaling spectrum, and integrate mutation profiling into clinical management will be pivotal in advancing personalized treatment and improving patient outcomes.
Acknowledgement: None.
Funding Statement: This research was funded by grants from the Cathay General Hospital in Taipei (CGH-MR-A11326 to C.-H.L.).
Author Contributions: The authors confirm contribution to the paper as follows: Writing—original draft preparation, Chung-Che Tsai, Chih-Hung Lin, Po-Chih Hsu, and Chan-Yen Kuo; writing, review and editing: Chung-Che Tsai, Chih-Hung Lin, Hsu-Hung Chang, Jin-Yin Chang, Tin-Yi Chu, Po-Chih Hsu, and Chan-Yen Kuo; literature compilation and systematic preparation of tables, or table compilation: Chung-Che Tsai, Chih-Hung Lin, Katherine Lin, Jia Hong Hubert Chen, Ying Jie Celia Chen, Ilyssa Ting-Ying Chang, Hsu-Hung Chang, Jin-Yin Chang, and Tin-Yi Chu; funding acquisition, Chih-Hung Lin. The detailed descriptions of the specific contributions made by these four authors are as follows: Katherine Lin: Conducted targeted literature searches on the molecular mechanisms discussed in Sections 2 and 3, extracted key findings, and assisted in summarizing supporting evidence for Tables 1 and 2; Jia Hong Hubert Chen: Compiled literature on HBx mutation-specific functions, cross-checked mutation-associated phenotypes, and assisted in the systematic organization of mutation-related content in Table 3; Ying Jie Celia Chen: Assisted in gathering references related to signaling pathways, verified citation accuracy, and contributed to the organization and validation of data entries for Tables 4 and 5; Ilyssa Ting-Ying Chang: Screened literature pertaining to noncoding RNA regulation, organized extracted data, and assisted in compiling and formatting the entries included in Tables 6 and 7. All authors fully acknowledge and accept complete academic responsibility for the research presented in this manuscript. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
Abbreviations
| a.a. | Amino acid |
| ABCB1 | ATP binding cassette subfamily B member 1 |
| AFP | α-Fetoprotein |
| Akt | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| APOBEC3B | Apolipoprotein B mRNA editing enzyme catalytic subunit 3B |
| ASCL1 | Achaete-scute family bHLH transcription factor 1 |
| ATF | Activating transcription factor |
| Bcl-xL | B-cell lymphoma-extra large |
| BRIX1 | Ribosome biogenesis protein BRX1 homolog |
| CAF | Cancer-associated fibroblast |
| Cav-1 | Caveolin-1 |
| CBP | CREB-binding protein |
| cccDNA | Covalently closed circular DNA |
| CD | Cluster of differentiation |
| CDC20 | Cell division cycle protein 20 |
| CHB | Chronic hepatitis B |
| CREB | cAMP response element-binding protein |
| CSC | Cancer stem cell |
| DMC | 2,5-Dimethylcelecoxib |
| DNTTIP2 | Deoxynucleotidyltransferase terminal interacting protein 2 |
| DUB | Deubiquitinating enzyme |
| E2F1 | E2F transcription factor 1 |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| ERK | Extracellular signal-regulated kinase |
| EZH2 | Enhancer of zeste homolog 2 |
| Fah | Fumarylacetoacetate hydrolase |
| FOXO1 | Forkhead box O1 |
| FRMD5 | FERM domain-containing protein 5 |
| Gab1 | GRB2-associated binding protein 1 |
| GPX4 | Glutathione peroxidase 4 |
| HBV | Hepatitis B virus |
| HBx | Hepatitis B virus X protein |
| HBxΔC | C-terminal truncated hepatitis B virus X protein |
| HBsAg | Hepatitis B surface antigen |
| HCC | Hepatocellular carcinoma |
| HSC | Hepatic stellate cell |
| HSF1 | Heat shock factor protein 1 |
| HSPA8 | Heat shock protein family A member 8 |
| HULC | HCC up-regulated long non-coding RNA |
| HUVEC | Human umbilical vein endothelial cell |
| IL | Interleukin |
| JAK | Janus kinase |
| JNK | c-Jun N-terminal kinase |
| let-7c | Lethal-7c microRNA |
| LLPS | Liquid-liquid phase separation |
| LONP1 | Lon peptidase 1, mitochondrial |
| LRP6 | Low-density lipoprotein receptor-related protein 6 |
| MALAT1 | Metastasis-associated lung adenocarcinoma transcript 1 |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MIF | Macrophage migration inhibitory factor |
| miRNA | MicroRNA |
| MLL1 | Myeloid-lymphoid leukemia protein 1 |
| mTOR | Mechanistic target of rapamycin |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NRAS | Neuroblastoma RAS viral oncogene homolog |
| PAI1 | Plasminogen activator inhibitor-1 |
| PD-L1 | Programmed death-ligand 1 |
| PI3K | Phosphoinositide 3-kinase |
| PINK1 | PTEN-induced putative kinase 1 |
| PrPC | Cellular prion protein |
| PSMC3 | Proteasome 26S subunit, ATPase 3 |
| PTEN | Phosphatase and tensin homolog |
| RBX1 | RING box protein 1 |
| RASSF1A | Ras association domain family 1 isoform A |
| RPF2 | Ribosome production factor 2 homolog |
| ROS | Reactive oxygen species |
| SHP2 | Src homology region 2-containing protein tyrosine phosphatase 2 |
| Skp2 | S-phase kinase associated protein 2 |
| SLC7A11 | Solute carrier family 7 member 11 |
| SMAD4 | SMAD family member 4 |
| SP1 | Specificity protein 1 |
| SPP1 | Secreted phosphoprotein 1 (osteopontin) |
| SPG21 | Spastic paraplegia 21 |
| STAT | Signal transducer and activator of transcription |
| ST8SIA6-AS1 | ST8SIA6 antisense RNA 1 |
| TAB2/3 | TAK1-binding protein 2/3 |
| TAK1 | TGF-beta activated kinase 1 |
| TAM | Tumor-associated macrophage |
| TBK1 | TANK-binding kinase 1 |
| TCF | T-cell factor |
| TGF-β | Transforming growth factor β |
| TFII-I | General transcription factor II-I |
| TFIIB | Transcription factor II B |
| TIB | Tumor immune barrier |
| TME | Tumor microenvironment |
| TNF-α | Tumor necrosis factor alpha |
| TRAF2/6 | TNF receptor-associated factor 2/6 |
| TRERNA1 | Transcriptional regulatory RNA 1 |
| TRPM7 | Transient receptor potential cation channel subfamily M member 7 |
| vWF | von Willebrand factor |
| VCPIP1 | Valosin-containing protein-interacting protein 1 |
| VEGF | Vascular endothelial growth factor |
| WDR75 | WD repeat domain 75 |
References
1. Abdelhamed W, El-Kassas M. Hepatocellular carcinoma recurrence: predictors and management. Liver Res. 2023;7(4):321–32. doi:10.1016/j.livres.2023.11.004. [Google Scholar] [PubMed] [CrossRef]
2. Russo FP, Zanetto A, Pinto E, Battistella S, Penzo B, Burra P, et al. Hepatocellular carcinoma in chronic viral hepatitis: where do we stand? Int J Mol Sci. 2022;23(1):500. doi:10.3390/ijms23010500. [Google Scholar] [PubMed] [CrossRef]
3. Li D, Hamadalnil Y, Tu T. Hepatitis B viral protein HBx: roles in viral replication and hepatocarcinogenesis. Viruses. 2024;16(9):1361. doi:10.3390/v16091361. [Google Scholar] [PubMed] [CrossRef]
4. Medhat A, Arzumanyan A, Feitelson MA. Hepatitis B x antigen (HBx) is an important therapeutic target in the pathogenesis of hepatocellular carcinoma. Oncotarget. 2016;12(24):2421–33. doi:10.18632/oncotarget.28077. [Google Scholar] [PubMed] [CrossRef]
5. Schollmeier A, Basic M, Glitscher M, Hildt E. The impact of HBx protein on mitochondrial dynamics and associated signaling pathways strongly depends on the hepatitis B virus genotype. J Virol. 2024;98(5):e00424–24. doi:10.1128/jvi.00424-24. [Google Scholar] [PubMed] [CrossRef]
6. Chae S, Ji JH, Kwon SH, Lee HS, Lim JM, Kang D, et al. HBxAPα/Rsf-1-mediated HBx-hBubR1 interactions regulate the mitotic spindle checkpoint and chromosome instability. Carcinogenesis. 2013;34(7):1680–8. doi:10.1093/carcin/bgt105. [Google Scholar] [PubMed] [CrossRef]
7. Hernández S, Álvarez-Astudillo F, Garrido D, Prieto C, Loyola A, Villanueva RA. Canonical and divergent N-terminal HBx isoform proteins unveiled: characteristics and roles during HBV replication. Biomedicines. 2021;9(11):1701. doi:10.3390/biomedicines9111701. [Google Scholar] [PubMed] [CrossRef]
8. Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol. 2004;78(23):12725–34. doi:10.1128/jvi.78.23.12725-12734.2004. [Google Scholar] [PubMed] [CrossRef]
9. Yun C, Um HR, Jin YH, Wang JH, Lee MO, Park S, et al. NF-κB activation by hepatitis B virus X (HBx) protein shifts the cellular fate toward survival. Cancer Lett. 2002;184(1):97–104. doi:10.1016/S0304-3835(02)00187-8. [Google Scholar] [PubMed] [CrossRef]
10. Villanueva RA, Loyola A. The intrinsically disordered region of HBx and virus-host interactions: uncovering new therapeutic approaches for HBV and cancer. Int J Mol Sci. 2025;26(8):3552. doi:10.3390/ijms26083552. [Google Scholar] [PubMed] [CrossRef]
11. Waris G, Huh KW, Siddiqui A. Mitochondrially associated hepatitis B virus X protein constitutively activates transcription factors STAT-3 and NF-κB via oxidative stress. Mol Cell Biol. 2001;21(22):7721–30. doi:10.1128/MCB.21.22.7721-7730.2001. [Google Scholar] [PubMed] [CrossRef]
12. Rahmani Z, Huh KW, Lasher R, Siddiqui A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J Virol. 2000;74(6):2840–6. doi:10.1128/jvi.74.6.2840-2846.2000. [Google Scholar] [PubMed] [CrossRef]
13. Schollmeier A, Glitscher M, Hildt E. Relevance of HBx for hepatitis B virus-associated pathogenesis. Int J Mol Sci. 2023;24(5):4964. doi:10.3390/ijms24054964. [Google Scholar] [PubMed] [CrossRef]
14. Ali A, Abdel-Hafiz H, Suhail M, Al-Mars A, Zakaria MK, Fatima K, et al. Hepatitis B virus, HBx mutants and their role in hepatocellular carcinoma. World J Gastroenterol. 2014;20(30):10238–48. doi:10.3748/wjg.v20.i30.10238. [Google Scholar] [PubMed] [CrossRef]
15. Ma J, Sun T, Park S, Shen G, Liu J. The role of hepatitis B virus X protein is related to its differential intracellular localization. Acta Biochim Biophys Sin. 2011;43(8):583–8. doi:10.1093/abbs/gmr048. [Google Scholar] [PubMed] [CrossRef]
16. Chong CK, Cheng CYS, Tsoi SYJ, Huang FY, Liu F, Fung J, et al. HBV X protein mutations affect HBV transcription and association of histone-modifying enzymes with covalently closed circular DNA. Sci Rep. 2020;10(1):802. doi:10.1038/s41598-020-57637-z. [Google Scholar] [PubMed] [CrossRef]
17. Villanueva RA, Loyola A. Pre- and post-transcriptional control of HBV gene expression: the road traveled towards the new paradigm of HBx, its isoforms, and their diverse functions. Biomedicines. 2023;11(6):1674. doi:10.3390/biomedicines11061674. [Google Scholar] [PubMed] [CrossRef]
18. Xu HZ, Liu YP, Guleng B, Ren JL. Hepatitis B virus-related hepatocellular carcinoma: pathogenic mechanisms and novel therapeutic interventions. Gastrointest Tumors. 2014;1(3):135–45. doi:10.1159/000365307. [Google Scholar] [PubMed] [CrossRef]
19. Kew MC. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J Gastroenterol Hepatol. 2011;26(s1):144–52. doi:10.1111/j.1440-1746.2010.06546.x. [Google Scholar] [PubMed] [CrossRef]
20. Ling LR, Zheng DH, Zhang ZY, Xie WH, Huang YH, Chen ZX, et al. Effect of HBx on inflammation and mitochondrial oxidative stress in mouse hepatocytes. Oncol Lett. 2020;19(4):2861–9. doi:10.3892/ol.2020.11404. [Google Scholar] [PubMed] [CrossRef]
21. Guo Q, Jin Y, Chen X, Ye X, Shen X, Lin M, et al. NF-κB in biology and targeted therapy: new insights and translational implications. Sig Transduct Target Ther. 2024;9(1):53. doi:10.1038/s41392-024-01757-9. [Google Scholar] [PubMed] [CrossRef]
22. Kim HR, Lee SH, Jung G. The hepatitis B viral X protein activates NF-κB signaling pathway through the up-regulation of TBK1. FEBS Lett. 2010;584(3):525–30. doi:10.1016/j.febslet.2009.11.091. [Google Scholar] [PubMed] [CrossRef]
23. Guo SP, Wang WL, Zhai YQ, Zhao YL. Expression of nuclear factor-κB in hepatocellular carcinoma and its relation with the X protein of hepatitis B virus. World J Gastroenterol. 2001;7(3):340–4. doi:10.3748/wjg.v7.i3.340. [Google Scholar] [PubMed] [CrossRef]
24. Liu Y, Zhang J, Zhai Z, Liu C, Yang S, Zhou Y, et al. Upregulated PrP(C) by HBx enhances NF-κB signal via liquid-liquid phase separation to advance liver cancer. npj Precis Oncol. 2024;8(1):211. doi:10.1038/s41698-024-00697-5. [Google Scholar] [PubMed] [CrossRef]
25. Kang HJ, Chung DH, Sung CO, Yoo SH, Yu E, Kim N, et al. SHP2 is induced by the HBx-NF-κB pathway and contributes to fibrosis during human early hepatocellular carcinoma development. Oncotarget. 2017;8(16):27263–76. doi:10.18632/oncotarget.15930. [Google Scholar] [PubMed] [CrossRef]
26. Feitelson MA, Arzumanyan A, Spector I, Medhat A. Hepatitis B x (HBx) as a component of a functional cure for chronic hepatitis B. Biomedicines. 2022;10(9):2210. doi:10.3390/biomedicines10092210. [Google Scholar] [PubMed] [CrossRef]
27. Hong J, Wu Y, Li M, Man KF, Song D, Koh SB. cAMP response element-binding protein: a credible cancer drug target. J Pharmacol Exp Ther. 2025;392(4):103529. doi:10.1016/j.jpet.2025.103529. [Google Scholar] [PubMed] [CrossRef]
28. Chen M, Liu Y, Yang Y, Qiu Y, Wang Z, Li X, et al. Emerging roles of activating transcription factor (ATF) family members in tumourigenesis and immunity: implications in cancer immunotherapy. Genes Dis. 2021;9(4):981–99. doi:10.1016/j.gendis.2021.04.008. [Google Scholar] [PubMed] [CrossRef]
29. Neuveut C, Wei Y, Buendia MA. Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol. 2010;52(4):594–604. doi:10.1016/j.jhep.2009.10.033. [Google Scholar] [PubMed] [CrossRef]
30. Cougot D, Wu Y, Cairo S, Caramel J, Renard CA, Lévy L, et al. The hepatitis B virus X protein functionally interacts with CREB-binding protein/p300 in the regulation of CREB-mediated transcription. J Biol Chem. 2007;282(7):4277–87. doi:10.1074/jbc.M606774200. [Google Scholar] [PubMed] [CrossRef]
31. Duan L, Wu R, Zhang X, Wang D, You Y, Zhang Y, et al. HBx-induced S100A9 in NF-κB dependent manner promotes growth and metastasis of hepatocellular carcinoma cells. Cell Death Dis. 2018;9(6):629. doi:10.1038/s41419-018-0512-2. [Google Scholar] [PubMed] [CrossRef]
32. Kang Y, Li W, Wei J, Yang L, Kang Y. Transcriptional regulation of tumor suppressor gene RASSF1A by HBx. Mol Cell Probes. 2025;82:102034. doi:10.1016/j.mcp.2025.102034. [Google Scholar] [PubMed] [CrossRef]
33. Chen C, Wang Z, Ding Y, Qin Y. Tumor microenvironment-mediated immune evasion in hepatocellular carcinoma. Front Immunol. 2023;14:1133308. doi:10.3389/fimmu.2023.1133308. [Google Scholar] [PubMed] [CrossRef]
34. Zhong J, Li Y, Liu Y, Qiao J, Wu Y, Kong X, et al. Hepatitis B virus X protein (HBx)-mediated immune modulation and prognostic model development in hepatocellular carcinoma. PLoS One. 2025;20(6):e0325363. doi:10.1371/journal.pone.0325363. [Google Scholar] [PubMed] [CrossRef]
35. Wang MD, Xiang H, Hong TY, Mierxiati A, Yan FH, Zhang L, et al. Integrated analysis of intratumoral biomarker and tumor-associated macrophage to improve the prognosis prediction in cancer patients. BMC Cancer. 2023;23(1):593. doi:10.1186/s12885-023-11027-6. [Google Scholar] [PubMed] [CrossRef]
36. Chen Z, Chen Y, Peng L, Wang X, Tang N. 2,5-dimethylcelecoxib improves immune microenvironment of hepatocellular carcinoma by promoting ubiquitination of HBx-induced PD-L1. J Immunother Cancer. 2020;8(2):e001377. doi:10.1136/jitc-2020-001377. [Google Scholar] [PubMed] [CrossRef]
37. Heindryckx F, Gerwins P. Targeting the tumor stroma in hepatocellular carcinoma. World J Hepatol. 2015;7(2):165–76. doi:10.4254/wjh.v7.i2.165. [Google Scholar] [PubMed] [CrossRef]
38. Rawal P, Siddiqui H, Hassan M, Choudhary MC, Tripathi DM, Nain V, et al. Endothelial cell-derived TGF-β promotes epithelial-mesenchymal transition via CD133 in HBx-infected hepatoma cells. Front Oncol. 2019;9:308. doi:10.3389/fonc.2019.00308. [Google Scholar] [PubMed] [CrossRef]
39. Liu Y, Xun Z, Ma K, Liang S, Li X, Zhou S, et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J Hepatol. 2023;78(4):770–82. doi:10.1016/j.jhep.2023.01.011. [Google Scholar] [PubMed] [CrossRef]
40. Huang XY, Li D, Chen ZX, Huang YH, Gao WY, Zheng BY, et al. Hepatitis B virus X protein elevates Parkin-mediated mitophagy through Lon Peptidase in starvation. Exp Cell Res. 2018;368(1):75–83. doi:10.1016/j.yexcr.2018.04.016. [Google Scholar] [PubMed] [CrossRef]
41. Swarnalatha M, Singh AK, Kumar V. Promoter occupancy of MLL1 histone methyltransferase seems to specify the proliferative and apoptotic functions of E2F1 in a tumour microenvironment. J Cell Sci. 2013;126(Pt 20):4636–46. doi:10.1242/jcs.126235. [Google Scholar] [PubMed] [CrossRef]
42. Mahboobnia K, Beveridge DJ, Yeoh GC, Kabir TD, Leedman PJ. microRNAs in hepatocellular carcinoma pathogenesis: insights into mechanisms and therapeutic opportunities. Int J Mol Sci. 2024;25(17):9393. doi:10.3390/ijms25179393. [Google Scholar] [PubMed] [CrossRef]
43. Zhu Y, Zhu Y, Deng Q, Liang X. Hepatitis B virus X protein promotes VWF-mediated HCC progression through ST8SIA6-AS1/miR-3150b-3p/ASCL1 axis. Eur J Pharmacol. 2025;991(Suppl. 1):177315. doi:10.1016/j.ejphar.2025.177315. [Google Scholar] [PubMed] [CrossRef]
44. Zhou P, Yao W, Liu L, Yan Q, Chen X, Wei X, et al. SPG21, a potential oncogene targeted by miR-128-3p, amplifies HBx-induced carcinogenesis and chemoresistance via activation of TRPM7-mediated JNK pathway in hepatocellular carcinoma. Cell Oncol. 2024;47(5):1757–78. doi:10.1007/s13402-024-00955-5. [Google Scholar] [PubMed] [CrossRef]
45. Khosravi M, Behboudi E, Razavi-Nikoo H, Tabarraei A. Hepatitis B virus X protein induces expression changes of miR-21, miR-22, miR-122, miR-132, and miR-222 in Huh-7 cell line. Arch Razi Inst. 2024;79(1):111–9. doi:10.32592/ARI.2024.79.1.111. [Google Scholar] [PubMed] [CrossRef]
46. Wu CS, Chien YC, Yen CJ, Wu JY, Bai LY, Yu YL. EZH2-mediated epigenetic silencing of tumor-suppressive let-7c/miR-99a cluster by hepatitis B virus X antigen enhances hepatocellular carcinoma progression and metastasis. Cancer Cell Int. 2023;23(1):199. doi:10.1186/s12935-023-03002-9. [Google Scholar] [PubMed] [CrossRef]
47. Niu LJ, Huang T, Wang L, Sun XF, Zhang YM. HBX suppresses PTEN to promote the malignant progression of hepatocellular carcinoma through mi-R155 activation. Ann Hepatol. 2022;27(3):100688. doi:10.1016/j.aohep.2022.100688. [Google Scholar] [PubMed] [CrossRef]
48. Yu G, Mu H, Zhou H, Fang F, Cui Y, Wu Q, et al. microRNA-361 suppresses the biological processes of hepatic stellate cells in HBV-relative hepatic fibrosis by NF-κB p65. Cells Dev. 2021;167:203711. doi:10.1016/j.cdev.2021.203711. [Google Scholar] [PubMed] [CrossRef]
49. Song W, Zheng C, Liu M, Xu Y, Qian Y, Zhang Z, et al. TRERNA1 upregulation mediated by HBx promotes sorafenib resistance and cell proliferation in HCC via targeting NRAS by sponging miR-22-3p. Mol Ther. 2021;29(8):2601–16. doi:10.1016/j.ymthe.2021.04.011. [Google Scholar] [PubMed] [CrossRef]
50. Zhang C, Liu P, Zhang C. Hepatitis B virus X protein upregulates alpha-fetoprotein to promote hepatocellular carcinoma by targeting miR-1236 and miR-329. J Cell Biochem. 2020;121(3):2489–99. doi:10.1002/jcb.29471. [Google Scholar] [PubMed] [CrossRef]
51. Liu Y, Feng J, Sun M, Yang G, Yuan H, Wang Y, et al. Long non-coding RNA HULC activates HBV by modulating HBx/STAT3/miR-539/APOBEC3B signaling in HBV-related hepatocellular carcinoma. Cancer Lett. 2019;454(Suppl 3):158–70. doi:10.1016/j.canlet.2019.04.008. [Google Scholar] [PubMed] [CrossRef]
52. He B, Peng F, Li W, Jiang Y. Interaction of lncRNA-MALAT1 and miR-124 regulates HBx-induced cancer stem cell properties in HepG2 through PI3K/Akt signaling. J Cell Biochem. 2019;120(3):2908–18. doi:10.1002/jcb.26823. [Google Scholar] [PubMed] [CrossRef]
53. Li R, Wang C, Xu K, Zhan Z, He S, Ren J, et al. Asiatic acid inhibits HBV cccDNA transcription by promoting HBx degradation. Virol J. 2024;21(1):268. doi:10.1186/s12985-024-02535-3. [Google Scholar] [PubMed] [CrossRef]
54. Ren F, Hu J, Dang Y, Deng H, Ren J, Cheng S, et al. Sphondin efficiently blocks HBsAg production and cccDNA transcription through promoting HBx degradation. J Med Virol. 2023;95(3):e28578. doi:10.1002/jmv.28578. [Google Scholar] [PubMed] [CrossRef]
55. Zhang Y, Li L, Cheng ST, Qin YP, He X, Li F, et al. Rapamycin inhibits hepatitis B virus covalently closed circular DNA transcription by enhancing the ubiquitination of HBx. Front Microbiol. 2022;13:850087. doi:10.3389/fmicb.2022.850087. [Google Scholar] [PubMed] [CrossRef]
56. Wang C, Niu W, Hua J, Zhao T, Feng H, Hao Z, et al. Spatiotemporal modulation of SMAD4 by HBx is required for cellular proliferation in hepatitis B-related liver cancer. Cell Oncol. 2022;45(4):573–89. doi:10.1007/s13402-022-00683-8. [Google Scholar] [PubMed] [CrossRef]
57. Wang Y, Zhao M, Zhao L, Geng Y, Li G, Chen L, et al. HBx-induced HSPA8 stimulates HBV replication and suppresses ferroptosis to support liver cancer progression. Cancer Res. 2023;83(7):1048–61. doi:10.1158/0008-5472.CAN-22-3169. [Google Scholar] [PubMed] [CrossRef]
58. Shoraka S, Hosseinian SM, Hasibi A, Ghaemi A, Mohebbi SR. The role of hepatitis B virus genome variations in HBV-related HCC: effects on host signaling pathways. Front Microbiol. 2023;14:1213145. doi:10.3389/fmicb.2023.1213145. [Google Scholar] [PubMed] [CrossRef]
59. Wu Q, Zhang L, Xu X, Zhang Y, Shi J, Lin X, et al. Hepatitis B virus X protein is stabilized by the deubiquitinating enzyme VCPIP1 in a ubiquitin-independent manner by recruiting the 26S proteasome subunit PSMC3. J Virol. 2022;96(13):e00611–22. doi:10.1128/jvi.00611-22. [Google Scholar] [PubMed] [CrossRef]
60. Pu R, Liu W, Zhou X, Chen X, Hou X, Cai S, et al. The effects and underlying mechanisms of hepatitis B virus X gene mutants on the development of hepatocellular carcinoma. Front Oncol. 2022;12:836517. doi:10.3389/fonc.2022.836517. [Google Scholar] [PubMed] [CrossRef]
61. Lin YH, Lai MW, Chu YD, Lin KH, Hsu CW, Chien RN, et al. HBx mutant-regulated RPL13AP25 mediates suboptimal virological response to entecavir? And HCC progression. Cancer Cell Int. 2025;25(1):223. doi:10.1186/s12935-025-03873-0. [Google Scholar] [PubMed] [CrossRef]
62. Jegal ME, Jung SY, Han YS, Kim YJ. C-terminal truncated HBx reduces doxorubicin cytotoxicity via ABCB1 upregulation in Huh-7 hepatocellular carcinoma cells. BMB Rep. 2019;52(5):330–5. doi:10.5483/BMBRep.2019.52.5.312. [Google Scholar] [PubMed] [CrossRef]
63. Mao X, Tey SK, Ko FCF, Kwong EML, Gao Y, Ng IO, et al. C-terminal truncated HBx protein activates caveolin-1/LRP6/β-catenin/FRMD5 axis in promoting hepatocarcinogenesis. Cancer Lett. 2019;444:60–9. doi:10.1016/j.canlet.2018.12.003. [Google Scholar] [PubMed] [CrossRef]
64. Siddiqui ZI, Farooqui SR, Azam SA, Afroz M, Wajid S, Parveen S, et al. A comparative study of hepatitis B virus X protein mutants K130M, V131I and KV130/131MI to investigate their roles in fibrosis, cirrhosis and hepatocellular carcinoma. J Viral Hepat. 2017;24(12):1121–31. doi:10.1111/jvh.12747. [Google Scholar] [PubMed] [CrossRef]
65. Hagiwara S, Nishida N, Park AM, Komeda Y, Sakurai T, Watanabe T, et al. Contribution of C1485T mutation in the HBx gene to human and murine hepatocarcinogenesis. Sci Rep. 2017;7(1):10440. doi:10.1038/s41598-017-10570-0.S. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools