
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
SOX11 Alleviates Osteoarthritis through Reducing Mitochondrial Dysfunction and Ferroptosis via Binding to the Promoter of NOX4
Department of Joint Surgery, Hunan Aerospace Hospital, Changsha, China
* Corresponding Author: Gang Yang. Email:
(This article belongs to the Special Issue: Modulation of Inflammation, Oxidative Stress, and Mitochondrial Function: Therapeutic Perspectives Across Diseases)
BIOCELL 2026, 50(4), 8 https://doi.org/10.32604/biocell.2026.074951
Received 22 October 2025; Accepted 15 January 2026; Issue published 21 April 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
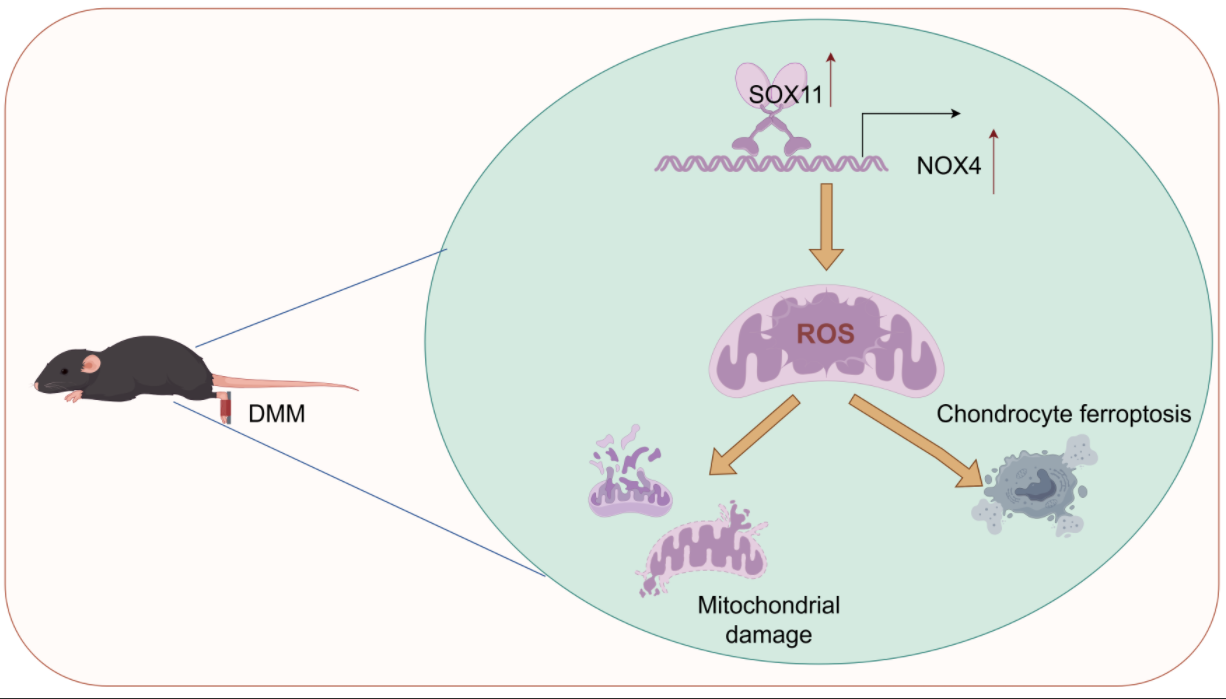
Objectives: Mitochondrial dysfunction and ferroptosis play crucial roles in osteoarthritis (OA), but the mechanisms remain unclear. This study aims to investigate the mechanism of sex-determining region Y-box transcription factor (SOX) 11/nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4) axis-mediated mitochondrial dysfunction and ferroptosis in OA. Methods: Destabilization of the medial meniscus (DMM) induced knee OA in mice. Chondrocytes were stimulated with IL-1β. Ferroptosis and mitochondrial function-related indicators were detected by immunofluorescence, 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1) staining, flow cytometry, quantitative real-time polymerase chain reaction (qRT-PCR), and Western blot. Results: OA mice had 4.4 and 1.1-fold increase in SOX11 and NOX4 proteins (p < 0.05). ChIP-qPCR and dual-luciferase reporter gene assays confirmed that SOX11 directly bound to the NOX4 promoter. Knocking SOX11 inhibited NOX4 and acyl-CoA synthetase long-chain family member 4 (ACSL4) levels in IL-1β-induced chondrocytes, promoted glutathione peroxidase 4 (GPX4) and solute carrier family 7 member 11 (SLC7A11) expression, and reduced Fe2+ concentration and cell death. Additionally, SOX11 knockdown inhibited lipid peroxidation and ROS production and malondialdehyde (MDA) level, increased mitochondrial membrane potential, GSH, and SOD levels. NOX4 overexpression interrupted the beneficial effects. In vivo experiments further validated that SOX11 knockdown reduced cartilage damage and Osteoarthritis Research Society International (OARSI) score, increased the transparent cartilage layer thickness, reduced calcified cartilage thickness, inhibited ferroptosis, and mitochondrial dysfunction. Conclusion: Our results indicate that SOX11 inhibition reduces mitochondrial dysfunction and ferroptosis in IL-1β-induced chondrocytes by downregulating NOX4, thereby alleviating OA. This finding provides new targets for OA treatment.Graphic Abstract
Keywords
Supplementary Material
Supplementary Material FileOsteoarthritis (OA) is a heterogeneous joint disorder characterized by pain and is a leading cause of disability and premature loss of work capacity [1]. OA significantly increases the global burden of disability, with its prevalence projected to rise by 74.9% by 2050 [2]. Pathologically, OA is characterized by progressive erosion of articular cartilage, leading to joint space narrowing, subchondral bone sclerosis, subchondral cyst formation, synovitis, and marginal osteophyte development [3]. Chondrocytes, as the sole cellular component of cartilage, are essential for cartilage maintenance and repair [4]. Extensive research indicates that chondrocyte apoptosis, a key event in OA pathogenesis, drives progressive cartilage degeneration [5]. However, due to the inability to determine the fundamental mechanism of disease onset, the severity of symptoms, and the complications of the disease, the treatment methods for OA remain inconsistent [6]. Therefore, developing novel therapeutic approaches is crucial for improving the clinical management of OA.
Ferroptosis is a programmed cell death form characterized by iron-dependent lipid peroxidation reactions [7]. Recent evidence indicates that the accumulation of iron ions accompanies the damaged areas of OA, and ferroptosis in chondrocytes plays a role in the pathogenesis of OA [8]. The cysteine/glutamate antiporter (System Xc−) is composed of solute carrier family 7 member 11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2), and is an important antioxidant system [9,10]. The activity of glutathione peroxidase 4 (GPX4) is highly dependent on the adequate intracellular glutathione (GSH) level, which is synthesized by System Xc− [11]. GPX4 oxidizes reduced GSH to oxidized glutathione and reduces toxic lipid peroxides (L-OOH) to corresponding alcohols, thereby reducing reactive oxygen species (ROS) production [12]. The downregulation of GPX4 increases the sensitivity of chondrocytes to oxidative stress and exacerbates extracellular matrix (ECM) degradation [13]. Mitochondrial dysfunction, characterized by impaired lipid metabolism and increased ROS production, leads to lipid peroxidation and ferroptosis [14]. Excessive intracellular iron accumulation induces mitochondrial dysfunction, leading to chondrocyte ferroptosis, which is a major factor contributing to cartilage damage in OA [15]. Targeting ferroptosis shows great potential in the treatment of OA [16]. Consequently, further exploration of the underlying molecular mechanisms linking mitochondrial dysfunction and ferroptosis in OA is imperative.
The sex-determining region Y-box transcription factor (SOX) C transcription factor superfamily, comprising SOX4, SOX11, and SOX12, is implicated in diverse physiological and pathological processes [17]. Notably, downregulation of SOXC gene expression in the synovial inner membrane of articular chondrocytes significantly reduces synovitis and articular cartilage erosion [18]. Compared with non-osteoarthritis subjects, the expression of SOX4 and SOX11 mRNA in the cartilage of osteoarthritis patients is increased [19,20]. Upregulation of miR-488-3p reduces the incidence of lipopolysaccharide (LPS)-induced chondrocyte damage by inhibiting SOX11 [21]. Tanshinone I inhibits interleukin (IL)-1β-induced chondrocyte apoptosis, inflammation and ECM degradation by down-regulating SOX11 [22]. In liver cancer tissues, the level of SOX11 is significantly positively correlated with immune infiltration, ferroptosis, and immune checkpoint genes [23]. However, it remains unclear whether SOX11 regulates mitochondrial dysfunction and ferroptosis in OA.
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4) is the main source of ROS [24]. NOX4 damages mitochondrial metabolism through lipid peroxidation caused by oxidative stress, thereby promoting ferroptosis [25]. Studies have shown that inhibiting the upregulation of NOX4 can alleviate mitochondrial dysfunction, effectively inhibit ferroptosis, and alleviate osteoarthritis [26]. Through the UCSC and JASPAR databases, we predicted that SOX11 is the transcription factor of NOX4. However, whether SOX11 affects OA by regulating mitochondrial dysfunction and ferroptosis through NOX4 remains unclear.
This study aims to investigate whether SOX11 exerts its effect in OA by directly regulating the expression of NOX4 through transcription, and to elucidate how the dysregulation of the SOX11/NOX4 pathway leads to mitochondrial dysfunction and ferroptosis in chondrocytes.
Twenty-four specific pathogen-free (SPF)-grade male C57BL/6 mice (twelve-week-old, 20–30 g) were purchased from Hunan SJA Laboratory Animal Co., Ltd. (Changsha, China). All animals were housed in individually ventilated cages, maintained in a controlled environment with a temperature of 23°C ± 1°C and a 12-h light/dark cycle. They had free access to water and food for 1 week before we performed destabilization of the medial meniscus (DMM) or sham surgery. According to variance analysis requirements, the within-group degrees of freedom (E) should be maintained between 10 and 20 to ensure statistical power [27]. E can be measured by the following formula: E = Total number of animals − Total number of groups. To investigate the effect of low expression of SOX11 in vivo, the mice were randomly divided into four groups, including Sham, OA, OA+sh-NC, and OA+sh-SOX11 (n = 6) groups. Knee OA was induced in mice via DMM as previously described [28]. Briefly, mice were anesthetized, and the right knee joint was aseptically sterilized. Subsequently, the medial joint capsule adjacent to the patellar tendon was carefully incised to expose the intercondylar region. The joint capsule and the overlying skin were sutured closed in layers. For the sham surgery group, the joint capsule was similarly opened but without disruption of the meniscus ligament. One week post-surgery, mice in OA+sh-NC and OA+sh-SOX11 groups were intra-articularly injected 1 × 109 pfu (8 μL) of short hairpin RNA (sh)-SOX11 lentivirus or corresponding negative control empty (sh-NC) lentivirus into the right knee joint, respectively. The mice were anesthetized via intraperitoneal injection of pentobarbital sodium (100 mg/kg) 8 weeks after surgery. Once the animals exhibited cessation of spontaneous breathing for 2–3 min and loss of the blink reflex, knee joint tissues were harvested for histopathologic analysis, Osteoarthritis Research Society International (OARSI) scoring, immunohistochemistry, and immunofluorescence. The Animal Experiment Ethics Committee for Biomedical Research, Hunan Normal University approved all animal experiment procedures (No.: 2025-637). All experimental procedures were conducted in accordance with institutional guidelines for the use of experimental animals.
The mouse knee joints were fixed in 4% paraformaldehyde at 4°C for 48 h, followed by decalcification in 10% EDTA (pH 7.4) for one month. Subsequently, the tissues were embedded in paraffin and serially sectioned at a thickness of 3 μm. The sections were baked at 62°C for 8 h, then subjected to dewaxing and rehydration procedures, and stained with either hematoxylin (Abiowell, AWI0001a, Changsha, China) and eosin (Abiowell, AWI0029a) or stained following Safranin-O/Fast Green kit (Abiowell, AWI0240a). After mounting with neutral balsam, the sections were examined under an optical microscope (MOTIC, BA210T, Xiamen, China).
According to histological analysis, tissue sections were obtained. Following immersion in 0.01 M sodium citrate antigen retrieval buffer (pH 6.0), microwave-mediated antigen retrieval was performed. After cooling at room temperature, the sections were then blocked with 1% periodate solution for 10 min at room temperature. Sections were subsequently washed three times in phosphate-buffered saline (PBS) (5 min per wash). After blocking, sections were incubated overnight at 4°C with the following primary antibodies against SOX11 (Bioss, bsm-60624R, 1:200, Rabbit, Beijing, China) and NOX4 (Proteintech, 14347-1-AP, 1:200, Rabbit, Chicago, IL, USA) or Rabbit IgG Isotype control (Proteintech, 98136-1-RR, 1:200). The samples were washed three times with PBS for 5 min each time. Sections were incubated with a ready-to-use HRP-conjugated polyclonal goat anti-rabbit IgG secondary antibody (Abiowell, AWI0629) at 37°C for 30 min. Following washing three times with PBS for 5 min each time, the sample was added with 50 μL of ready-to-use chromogenic reagent DAB solution (Abiowell, AWB0175) and incubated at room temperature for 30 s. The reaction time was controlled under an optical microscope (MOTIC, BA210T), and distilled water was used for washing to terminate the reaction. After three additional PBS washes (5 min per wash), cell nuclei were counterstained with hematoxylin for 10 min. Sections were observed and analyzed using an optical microscope (MOTIC, BA210T). For each sample, five random fields of view were selected under an optical microscope (MOTIC, BA210T). Positive expression analysis was performed by calculating the ratio of cumulative optical density from the positive expression area to the sample area within each field (Media Cybernetics, Image-Pro-Plus 6.0, Silver Spring, MD, USA). Each experiment is independently repeated three times.
Sections were obtained by referring to the histological analysis methods. Microwave-mediated antigen retrieval was performed. The cells were processed as follows: fixation in 4% paraformaldehyde (30 min), permeabilization with 0.3% Triton X-100 (30 min), and blocking with 5% bovine serum albumin (BSA, Abiowell, AWI0120a) at 37°C for 90 min. Sections were incubated overnight at 4°C with primary antibodies GPX4 (AWA11352, 1:200, Rabbit, Abiowell). Sections were incubated with secondary antibody, CoraLite488-conjugated Affinipure Goat Anti-Rabbit IgG (H+L) (Abiowell, AWS0005a, 1:200), at 37°C for 60 min. DAPI working solution (Abiowell, AWC0293a, 5 μg/mL) was nucleated for 10 min at 37°C. The sections were mounted in buffered glycerin and visualized using a fluorescence microscope (MOTIC, BA210T). For each sample, five random fields of view were selected. Positive expression analysis was performed by calculating the ratio of cumulative optical density from the positive expression area within each field (Media Cybernetics, Image-Pro-Plus 6.0). Each experiment is independently repeated three times.
2.5 Isolation and Culture of Chondrocytes
The tibiae and femora of P3-P5 neonatal mice were carefully dissected and sectioned into fragments approximately 1 mm3 in size. The harvested tissues were initially subjected to digestion using 0.25% trypsin at 37°C for 10 min. Subsequently, they were digested with 0.2% type II collagenase and then placed in an air-oscillating incubator (Thermo Fisher Scientific MaxQ4000). The samples were incubated at a rotation speed of 250 rpm and a temperature of 37°C for 8 h. Following digestion, the primary chondrocytes were isolated and seeded onto culture plates. Mycoplasma contamination testing confirmed the absence of contaminants in the supernatant of primary murine articular chondrocytes. Cells were plated at a density of 5 × 104 cells per well in a six-well plate. These cells were cultured in DMEM/F12 medium (Gibco, 11320033, Shanghai, China), which was supplemented with 1% penicillin-streptomycin solution (Gibco, 15140148) and 10% fetal bovine serum (FBS, Gibco, 10099141). The culture environment was maintained at 37°C with a 5% CO2 atmosphere. The primary chondrocytes were treated with 10 ng/mL IL-1β (Proteintech, 211-11B) at 37°C for 24 h [29]. Before the IL-1β treatment, cells were cultured in DMEM/F12 medium with a low serum condition (containing 1% FBS) and 1% penicillin-streptomycin solution. To ensure the completeness of the phenotype, we used the third-generation chondrocytes.
Chondrocytes were plated at a density of 5 × 104 cells per well in a six-well plate. Chondrocytes were cultured in DMEM/F12 medium supplemented with 1% penicillin-streptomycin solution and 10% fetal bovine serum at 37°C with a 5% CO2 atmosphere. Transfection was performed when cells reached 70%–80% confluence. In a 1-mL transfection system, chondrocytes were transfected with either small interfering RNA targeting SOX11 (si-SOX11; 5 μL, 100 nM, HG-SM009234, HonorGene, Changsha, China), NOX4 overexpression plasmid (oe-NOX4; 3 μg, HG-MO015760, HonorGene), or their corresponding negative controls (si-NC/oe-NC) using Lipofectamine 2000 transfection reagent (5 μL) according to the instructions. The transfection groups were designated as: si-NC, si-SOX11, si-SOX11+oe-NC, and si-SOX11+oe-NOX4. The sequences of the targeted siRNAs used in the study are as follows: si-NC: 5′-TAGGTATCGGATGGAGTTACC-3′; si-SOX11#1: 5′-GGCTCTACTACAGCTTCAAGA-3′; si-SOX11#2: 5′-GAGTAGTTTTAAACAGATTTT-3′.
2.7 Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Chondrocytes were seeded in a six-well plate at a density of 5 × 104 cells per well. Cells were lysed in TRIzol reagent (Thermo Fisher, 15596026, Waltham, MA, USA) for total RNA extraction. cDNA was then synthesized using an mRNA reverse transcription kit (CWBIO, CW2569, Beijing, China). The cDNA template was subjected to qRT-PCR amplification with the UltraSYBR Mixture (CWBIO, CW2601). The 30 μL qPCR reaction mixture contained: 5 μL template DNA (or input control), 1 μL Primer R, 1 μL Primer F, 8 μL ddH2O, and 15 μL 2× SYBR GREEN PCR Master Mix. The thermal cycling conditions were: initial denaturation at 95°C for 10 min; followed by 40 cycles of 95°C for 15 s and 60°C for 30 s. mRNA expression levels were calculated by the 2-ΔΔCt method, with β-actin serving as the internal control. Primer sequences are provided in Table 1.

Total proteins were extracted from either 5 × 104 cells or 25 mg tissues using RIPA lysis buffer (Beyotime, P0013B, Shanghai, China) containing protease inhibitors (583794, Jintai Hongda Biotechnology Co., Ltd., Beijing, China). Following 30 min incubation on ice, lysates were centrifuged at 12,000× g for 15 min at 4°C, and the supernatants were collected for subsequent analysis. The total protein was quantified via the BCA assay kit (Beyotime, P0012S). Proteins (20 μL) were then separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred onto nitrocellulose membranes. The membranes were blocked with 5% (w/v) skimmed milk powder at room temperature for 90 min, followed by overnight incubation at 4°C with primary antibodies or negative control. Subsequently, the membranes were incubated with HRP Goat anti-Mouse IgG (H+L) Secondary Antibody (1:5000, Abiowell, AWS0001) or HRP Goat anti-Rabbit IgG (H+L) Secondary Antibody (1:5000, Abiowell, AWS0002) for 90 min. β-actin served as the internal reference protein. Detailed information regarding the antibodies employed is provided in Table 2. For visualization, the membranes were incubated with ECL chemiluminescence reagent (K-12045-D50, Advansta, Menlo Park, CA, USA) for 1 min, and images were captured using a ChemiScope6100 gel imaging system (CLINX, Shanghai, China). The resulting exposed image bands were subjected to densitometric analysis using Quantity One professional grayscale analysis software (version 4.6.8, BIO-RAD, Hercules, CA, USA).

2.9 Chromatin Immunoprecipitation (ChIP) Assays
The ChIP assays followed the protocol of the ChIP Kit (Abcam, ab500). The cells (3 × 106) were cultured in a 10 cm petri dish and crosslinked with 1.1% formaldehyde (25 μL), Buffer A (45 μL), and PBS (0.6 mL) for 10 min at room temperature. The reaction was quenched with 65 μL glycine (1.25 M) at room temperature for 5 min. Cells were harvested by centrifugation at 500× g for 5 min at 4°C. The pellet was washed once with 1 mL of ice-cold PBS and centrifuged again under the same conditions. The washed cell pellet was sequentially resuspended in 1 mL of Buffer B and then 1 mL of Buffer C, with each incubation step performed at room temperature for 10 min. After each resuspension, the mixture was centrifuged at 500× g for 5 min and the supernatant discarded. The resulting nuclear pellet was resuspended in a mixture of 4 μL 25× PI Mix and 96 μL Buffer D. Chromatin was sheared by ultrasonication (XIAO MEI CHAO SHENG, XM-250T, Kunshan, Jiangsu, China) using the following parameters: 4 s ON/6 s OFF pulses, for a total sonication time of 60 s at 20% power output. The sonicated lysate was centrifuged at 14,000× g for 5 min at 4°C. The supernatant, containing the fragmented chromatin, was collected. A 20 μL aliquot of supernatant was mixed with 100 μL of ddH2O, followed by the addition of 100 μL of DNA purifying solution and thorough mixing. The mixture was incubated at 98°C for 10 min, followed by 20 min at room temperature. After centrifugation at 10,000 × g for 10 s, 1 μL proteinase K was added and vortexed briefly (5 s). Subsequent incubations were performed at 55°C for 30 min and then 98°C for 10 min. The mixture was finally centrifuged at 14,000× g for 1 min at room temperature. The supernatant was transferred to a new 1.5 mL tube. Fragment size distribution was analyzed by electrophoresis on a 1.5% agarose gel using 10 μL of this sample. Each ChIP reaction used 1 μg fragmented chromatin dissolved in 1 mL 1× ChIP buffer and vortexed for 5 s. The ChIP sample was incubated with either 2 μL IgG antibody (Merck Millipore, USA) or SOX11 polyclonal antibody (Thermo Fisher, PA5-47557) at 4°C overnight. The washed agarose beads (40 μL) were resuspended in 1× ChIP buffer (5.3 mL). A 120 μL aliquot of the bead suspension was mixed with 1 mL 1× ChIP buffer, centrifuged at 500× g for 5 min at 4°C, and the supernatant was removed. Meanwhile, the ChIP sample was centrifuged at 14000× g for 5 min at 4°C. The resulting supernatant was transferred to a fresh tube containing the pre-blocked agarose beads and incubated at 4°C for 30–60 min under constant rotation. Following incubation, the mixture was centrifuged at 500× g for 3 min, and the supernatant was discarded. The bead-bound immune complexes were subsequently washed three times with 1× ChIP buffer. The washed agarose beads were resuspended with 100 μL DNA purification slurry at 98°C for 10 min, followed by a 20-min incubation at room temperature, and centrifuged at 10,000× g for 10 s. Proteinase K (1 μL) was added to the obtained sample, vortexed for 5 s, incubated sequentially at 55°C for 30 min and then 98°C for 10 min, and centrifuged at 14000× g for 1 min. Then, 70 μL the clarified supernatant was carefully transferred to a new 1.5 mL microcentrifuge tube. The purified DNA products were tested by qRT-qPCR amplification with the UltraSYBR Mixture. The 30 μL qPCR reaction mixture contained: 5 μL template DNA (or input control), 1 μL Primer R, 1 μL Primer F, 8 μL ddH2O, and 15 μL 2× SYBR GREEN PCR Master Mix. The thermal cycling conditions were: initial denaturation at 95°C for 10 min; followed by 40 cycles of 95°C for 15 s and 60°C for 30 s. Primer sequences are provided in Table 1.
2.10 Dual-Luciferase Reporter Gene
The potential transcriptional binding sites of SOX11 and NOX4 were identified using the Jaspar database (https://jaspar.elixir.no/). The wild-type (WT) promoter region upstream of the NOX4 gene was amplified by PCR, and specific mutations (MUT) were introduced using site-directed mutagenesis techniques. Both the WT and MUT NOX4 promoter fragments were cloned into pGL3-Basic luciferase reporter vectors. HEK 293 T cells (HonorGene, HG-NC071) were maintained in DMEM medium in a six-well plate at a density of 5 × 104 cells per well. For luciferase reporter assays, HEK 293 T cells were transfected using Lipofectamine 2000 Transfection Kit (5 μL/well, Invitrogen, 11668-019, Carlsbad, CA, USA) according to the manufacturer’s instructions. In each experiment, pGL3-Basic-NOX4-WT (2 μg) or pGL3-Basic-NOX4-MUT (2 μg) reporter construct were transfected separately into distinct wells, along with 0.2 μg Renilla luciferase control plasmid (pRL-TK, HonorGene) and either 2 μg si-SOX11 or si-NC plasmid. Following 48 h of incubation at 37°C under 5% CO2, luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega, GloMax 20/20, Madison, WI, USA) in strict accordance with the manufacturer’s instructions. The firefly luciferase luminescence activity was normalized to the control Renilla luciferase activity. The experiment was independently repeated three times, representing biological replicates.
2.11 Biochemical Kit Detection
The concentration of malondialdehyde (MDA), superoxide dismutase (SOD), glutathione (GSH), and Fe2+ in cell lysates/tissues was measured using the MDA assay kit (Jiancheng, A003-1-2, Nanjing, Jiangsu, China), SOD kit (Jiancheng, A001-3-2), microreduced GSH assay kit (Jiancheng, A006-2-1), and an iron ion assay kit (Jianglaibio, JL-T1255, Shanghai, China). The determination procedure was based on the kit protocol.
2.12 Lipid Peroxidation Measurement
Chondrocytes were seeded in a six-well plate at a density of 5 × 104 cells per well. Following trypsinization, cells were harvested by centrifugation at 1000× g for 5 min at 4°C. Following resuspension in fresh culture medium, the cells were incubated with 5 μM C11-BODIPY (Invitrogen, D3861) at 37°C for 30 min in the dark. Lipid peroxidation was subsequently quantified using flow cytometry (Beckman Coulter, A00-1-1102, Shanghai, China) under 581 nm excitation wavelength, 591 nm emission wavelength.
2.13 Reactive Oxygen Species (ROS) Measurement
Chondrocytes were seeded in a six-well plate at a density of 5 × 104 cells per well. Following treatment, cells were detached with 0.25% (w/v) trypsin-EDTA solution (Thermo Fisher) and stained with 10 µM DCFH-DA (Beyotime, S0033S) diluted in serum-free culture medium at 37°C for 30 min. ROS levels were quantified by flow cytometry (Beckman Coulter, A00-1-1102). The maximum excitation wavelength is 488 nm, and the maximum emission wavelength is 525 nm.
2.14 5,5′,6,6′-Tetrachloro-1,1′,3,3′-Tetraethyl-Imidacarbocyanine-Cyanine Iodide (JC-1) Staining
Chondrocytes were seeded in a six-well plate at a density of 5 × 104 cells per well. Following incubation with 0.5 mL 1× JC-1 staining solution (Beyotime, #C2006) at 37°C for 10 min, cells were washed three times with JC-1 staining buffer. Flow cytometric analysis was subsequently performed, with the mitochondrial membrane potential (ΔΨm) being quantified through fluorescence ratio analysis. The red/green fluorescence intensity ratio (H1-UL aggregates to H1-UR monomers) served as the indicator of JC-1 polarization status. The maximum excitation wavelength of the JC-1 monomer is 514 nm, and the maximum emission wavelength is 529 nm; the maximum excitation wavelength of the JC-1 aggregates is 585 nm, and the maximum emission wavelength is 590 nm.
Chondrocytes were seeded in a six-well plate at a density of 5 × 104 cells per well. According to the manufacturer’s protocol, cell death was assessed using a live/dead cell staining kit (KeyGEN Bio TECH, KGAF001, Nanjing, China). Briefly, cells were incubated with 2 μM fluorescein AM and 8 μM propidium iodide at room temperature for 30 min. The percentage of cell death was quantified using flow cytometry (Beckman Coulter, A00-1-1102).
All data are presented as the mean ± standard deviation. Statistical analyses were performed using GraphPad Prism 8.0 software (GraphPad Software Inc., San Diego, CA, USA). For comparisons between two groups, an unpaired two-tailed Student’s t-test was utilized. For multiple group comparisons, one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test was employed. A p-value less than 0.05 was considered statistically significant.
3.1 The Expressions of SOX11 and NOX4 Were Upregulated in OA Mice
To verify the pathological features of the OA mouse model, we employed H&E staining, Safranin-O/Fast Green staining, and molecular detection [matrix metalloproteinase 13 (MMP13) and Collagen II levels]. In the Sham group, the cartilage exhibited a distinct layered structure (surface layer, middle layer, and deep layer) and orderly cell arrangement, characteristic of normal cartilage tissue. In contrast, the OA group displayed obvious surface cartilage damage, indicative of chondrocyte loss and matrix fibrosis. This damage was further quantified by an elevated OARSI score (Fig. 1A). Compared with the Sham group, the thickness of the transparent cartilage layer in the OA group was reduced, while the calcified cartilage thickness increased (Fig. 1B). This change strongly suggested the massive loss of proteoglycans and the occurrence of cartilage calcification. SOX11 and NOX4 expression in the knee joint tissue of the OA group was increased relative to the Sham group (Fig. 1C–E). The immunohistochemical negative control is presented in supplementary Fig. S1A. Additionally, the OA model induced an increase in MMP13 levels and a decrease in Collagen II levels (Fig. 1E). Negative controls of Western blot are shown in supplementary Fig. S1B. Our results indicate that the expressions of SOX11 and NOX4 are upregulated in OA mice, suggesting that they may serve as biomarkers for OA.

Figure 1: Elevated expression of sex-determining region Y-box transcription factor (SOX) 11 and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4) in OA mice. (A) Hematoxylin and eosin (H&E) staining was applied to show morphological changes in knee joint tissue and the Osteoarthritis Research Society International (OARSI) score. Scale bar: 100 μm or 25 μm. (B) Safranin-O/Fast Green staining was applied for assessing the degree of knee joint tissue damage. Scale bar: 100 μm or 25 μm. (C) Immunohistochemical analysis of SOX11 and NOX4 expression. Scale bar: 100 μm or 25 μm. (D) Quantitative real-time polymerase chain reaction (qRT-PCR) and (E) Western blot analysis of SOX11, NOX4, matrix metalloproteinase 13 (MMP13), and Collagen II expression. n = 3.
3.2 SOX11 Enhanced the Transcriptional Activity of NOX4 by Directly Binding to the Promoter of NOX4
Subsequently, the possible regulatory mechanisms of SOX11 and NOX4 in chondrocytes were analyzed in vitro. Compared with the si-NC group, SOX11 expression in the si-SOX11#1 and si-SOX11#2 groups were decreased, confirming the efficiency of si-SOX11 transfection (Fig. 2A,B). SOX11 knockdown reduced NOX4 expression in chondrocytes (Fig. 2A,B). ChIP-qPCR detection revealed that the binding efficiency of SOX11 to the promoter region of NOX4 in the SOX11 knockdown group was lower than that in the si-NC group (Fig. 2C). The dual-luciferase reporter gene system showed that the luciferase activity of the wild-type (WT) NOX4 promoter group decreased after SOX11 knockdown, while the mutant (MUT) promoter group showed no significant change (Fig. 2D). Our results confirm that SOX11 promotes NOX4 transcription in chondrocytes by directly binding to its promoter region.

Figure 2: SOX11 enhances the transcriptional activity of NOX4 by directly binding to the promoter of NOX4. (A) qRT-PCR and (B) Western blot analyses of SOX11 and NOX4 expression. (C) ChIP-qPCR analysis of the binding strength of SOX11 to the promoter region of NOX4. (D) Dual-luciferase reporter gene assay to detect the binding situation of SOX11 to the promoter region of NOX4. n = 3.
3.3 Down-Regulation of SOX11 Inhibits Ferroptosis in IL-1β-Induced Chondrocyte through NOX4
Emerging evidence establishes SOX11′s regulatory role in ferroptosis mechanisms [23]. Next, the SOX11 effect on ferroptosis in IL-1β-induced chondrocytes through regulating NOX4 was investigated. IL-1β stimulation upregulated SOX11, NOX4, and MMP13 levels in chondrocytes, while also causing a decrease in Collagen II levels (Fig. 3A,B). Knockdown of SOX11 inhibited NOX4 level in IL-1β-induced chondrocytes. When NOX4 was overexpressed, this inhibitory effect was reversed (Fig. 3C,D), confirming that SOX11 exerts its effect by positively regulating NOX4. IL-1β treatment upregulated ACSL4 level in chondrocytes and downregulated GPX4 and SLC7A11 levels. This phenomenon was reversed by knocking down SOX11. NOX4 overexpression eliminated these protective effects (Fig. 3E,F). Negative controls of Western blot are shown in Fig. S1B. SOX11 knockdown reduced the Fe2+ concentration and cell death in chondrocytes subjected to IL-1β, which was counteracted by NOX4 overexpression (Fig. 3G,H). Immunofluorescence results show that SOX11 knockdown increased GPX4 expression in chondrocytes subjected to IL-1β. NOX4 overexpression reversed this phenomenon (Fig. 3I). Our results indicate that knockdown of SOX11 reduces ferroptosis in chondrocytes subjected to IL-1β by down-regulating NOX4.

Figure 3: SOX11 down-regulation reduces ferroptosis in chondrocytes subjected to interleukin (IL)-1β via NOX4. (A) qRT-PCR and (B) Western blot analysis of SOX11, NOX4, MMP13, and Collagen II expression. (C) qRT-PCR and (D) Western blot analysis of SOX11 and NOX4 expression. (E) qRT-PCR and (F) Western blot analysis of acyl-CoA synthetase long-chain family member 4 (ACSL4), glutathione peroxidase 4 (GPX4), and solute carrier family 7 member 11 (SLC7A11) expression. (G) Fe2+ concentration was detected using a biochemical kit. (H) Cell death was assessed using a live/dead cell staining kit. (I) Immunofluorescence for GPX4 expression. Scale bar: 25 μm. n = 3.
3.4 Knocking down SOX11 Alleviates Mitochondrial Dysfunction in Chondrocytes Subjected to IL-1β through NOX4
Then, we further explored the SOX11 role in regulating NOX4 to exert its effect on mitochondrial dysfunction in chondrocytes subjected to IL-1β. Knocking down SOX11 inhibited lipid peroxidation in chondrocytes subjected to IL-1β. NOX4 overexpression reversed the downward trend of lipid peroxidation (Fig. 4A). Knocking down SOX11 increased the mitochondrial membrane potential in chondrocytes subjected to IL-1β, and this effect was counteracted by NOX4 overexpression (Fig. 4B). Additionally, low expression of SOX11 reduced ROS production and MDA levels in chondrocytes subjected to IL-1β, while increasing GSH and SOD levels. When NOX4 was overexpressed, these protective effects brought about by low expression of SOX11 were eliminated (Fig. 4C,D). Our results indicate that knocking down SOX11 alleviates mitochondrial dysfunction in chondrocytes subjected to IL-1β through NOX4.


Figure 4: SOX11 knockdown alleviates mitochondrial dysfunction in chondrocytes subjected to IL-1β through NOX4. (A) Flow cytometry was used to detect intracellular lipid peroxidation levels. (B) JC-1 was used to measure mitochondrial membrane potential. (C) Flow cytometry was applied for detecting reactive oxygen species (ROS) levels. (D) Biochemical kits were used to assess glutathione (GSH), superoxide dismutase (SOD), and malondialdehyde (MDA) levels. n = 3.
3.5 Reducing SOX11 Level Alleviates OA through Decreasing Ferroptosis and Mitochondrial Dysfunction
Finally, in vivo experiments were conducted to investigate SOX11 effects on ferroptosis and mitochondrial dysfunction in OA mice. Immunohistochemical results showed that SOX11 knockdown reduced SOX11 and NOX4 expression in the knee joint tissues of the OA group (Fig. 5A), and negative controls is presented in Fig. S1C. H&E staining results show that the Sham group clearly displayed the layered structure of cartilage (surface layer, middle layer, and deep layer) and cell arrangement, demonstrating the typical characteristics of normal cartilage tissue. However, in the OA group, the surface of the cartilage showed obvious damage, which directly indicated the loss of chondrocytes and matrix fibrosis. At the same time, the OARSI score was increased. Knockdown of SOX11 alleviated the cartilage damage induced by OA and reduced the OARSI score (Fig. 5B). SOX11 knockdown increased the thickness of the transparent cartilage layer in the knee joint tissues of OA model mice and reduced the thickness of calcified cartilage (Fig. 5C). These results indicate that SOX11 knockdown alleviates OA. In addition, SOX11 knockdown increased GPX4 level in the knee joint tissues of OA model mice (Fig. 6A) and mitochondrial membrane potential (Fig. 6B), reduced ROS production, MDA levels, and Fe2+ concentration, and increased GSH and SOD levels (Fig. 6C–E). Our results show that SOX11 knockdown alleviates OA through reducing ferroptosis and mitochondrial dysfunction.

Figure 5: Lowering SOX11 level alleviates OA. (A) Immunohistochemical analysis of SOX11 and NOX4 expression. Scale bar: 100 or 25 μm. (B) The morphological changes of knee joint tissues were observed by H&E staining and the OARSI score. Scale bar: 100 or 25 μm. (C) The degree of tissue damage in the knee joint was assessed using Safranin-O/Fast Green staining. Scale bar: 100 or 25 μm. n = 3.

Figure 6: Down-regulation of SOX11 reduces ferroptosis and mitochondrial dysfunction in OA mice. (A) Immunofluorescence detection of GPX4 expression. (B) JC-1 assessment of mitochondrial membrane potential. (C) Flow cytometry detection of ROS levels. (D) Biochemical kits for evaluating GSH, SOD, and MDA levels. (E) Biochemical kits for detecting Fe2+ concentration. n = 3.
OA is a common age-related disease characterized by dysregulation of extracellular matrix metabolism, lipid metabolism, and upregulation of the pro-inflammatory senescence-associated secretory phenotype (SASP) (such as IL1β, IL6, TNFα, MMP3, MMP13) [30,31]. Targeting ferroptosis in OA treatment is considered an important therapeutic approach [32]. Elucidating the molecular mechanisms underlying ferroptosis in OA pathogenesis is therefore critically significant. Here, we found that SOX11 and NOX4 were upregulated in the OA model mice. Mechanistic studies demonstrated that SOX11 enhanced the transcriptional activity of NOX4 in chondrocytes by directly binding to its promoter. In vitro results suggested that knockdown of SOX11 reduced ferroptosis and mitochondrial dysfunction in chondrocytes subjected to IL-1β by downregulating NOX4. The in vivo results further indicated that SOX11 knockdown alleviated cartilage damage and reduced the OARSI score, increased the thickness of the transparent cartilage layer, decreased the thickness of calcified cartilage, inhibited ferroptosis and mitochondrial dysfunction, and thereby alleviated OA.
Transcription factors are pivotal in preserving the homeostasis of normal tissues, with cartilage and bone being notable examples [33]. By binding to specific genomic DNA regions, these factors orchestrate the transcription of target genes, influencing the pathological processes within chondrocytes, subchondral bone, and synovial tissue, thereby modulating the initiation and progression of OA [34]. Consequently, targeted intervention in the upstream and downstream signaling networks associated with transcription factors holds considerable promise for the prevention and therapeutic management of OA [35]. We found that SOX11 was upregulated in the OA model mice, which was consistent with previous studies [21]. However, the precise molecular mechanisms underlying SOX11’s role in OA pathogenesis await further comprehensive elucidation. Similarly, NOX4 was upregulated in the OA model mice. Mechanistic analyses revealed that SOX11 directly bound to the NOX4 promoter in chondrocytes, enhancing its transcription and expression. These findings indicate that SOX11 might affect OA progression through direct transcriptional regulation of NOX4.
Emerging evidence highlights the pivotal role of ferroptosis in regulating the activity of chondrocytes, ECM degradation, and synovial inflammation, thereby exacerbating the progression of OA [36]. Elevated iron levels in joint tissues of OA patients may trigger ferroptosis in chondrocytes by enhancing oxidative stress and lipid peroxidation [37]. Furthermore, the pathogenesis of OA is associated with diminished expression levels of critical ferroptosis regulatory proteins, specifically GPX4 and SLC7A11, leading to enhanced ferroptosis in cartilage [38]. During ferroptosis, GPX4 uses reduced GSH to convert phospholipid peroxides into lipols and inhibits ferroptosis [39]. ACSL4 esterifies arachidonic acid and adrenal acid to form phosphatidylethanolamine, promoting the accumulation of lipid peroxides and ferroptosis in chondrocytes [40]. In mild OA patients, using ferroptosis inhibitors such as Ferrostatin-1 can significantly upregulate the expression of GPX4 and SLC7A11, while downregulating ACSL4 expression and MDA levels, and increasing mitochondrial membrane potential [41]. Recent evidence indicates that oxidative stress and mitochondrial dysfunction not only induce ferroptosis but also trigger chondrocyte senescence, promoting the release of pro-inflammatory SASP that exacerbates cartilage degeneration [42,43]. Inhibiting oxidative stress alleviated chondrocyte senescence and OA by improving mitochondrial function [44]. NOX4 plays a key role in the ROS-mediated lipid peroxidation process in mitochondrial damage and ferroptosis [45]. In IL-1β-induced chondrocytes, we observed that SOX11 knockdown downregulated NOX4, ACSL4, and MMP13. Concurrently, it enhanced Collagen II, GPX4, and SLC7A11 expression, reduced intracellular Fe²+ concentration, and decreased the cell death rate. Overexpression of NOX4 prevented the beneficial effects brought about by the low expression of SOX11. Our results indicate that knockdown of SOX11 reduced ferroptosis in chondrocytes subjected to IL-1β by downregulating NOX4. Excessive iron can induce mitochondrial dysfunction and cell death through ROS production, thereby promoting the expression of OA metabolic markers [46]. Ferrostatin-1, a ferroptosis inhibitor, has shown potential in reducing oxidative stress, restoring mitochondrial integrity, and reducing lipid peroxidation damage [47]. Astaxanthin reduces the levels of ROS, lipid peroxidation, and iron in chondrocytes subjected to IL-1β, promotes SLC7A11 and GPX4 expression, inhibits ferroptosis and mitochondrial dysfunction, and thereby alleviates OA [48]. We found that SOX11 knockdown inhibited lipid peroxidation and ROS production, and MDA level in chondrocytes subjected to IL-1β, increased mitochondrial membrane potential, GSH, and SOD levels. Overexpression of NOX4 reversed these protective effects of SOX11 knockdown. Our results indicate that SOX11 knockdown could reduce NOX4 expression and alleviate mitochondrial dysfunction in IL-1β-induced chondrocytes. The in vivo experiment results further confirm that knockdown of SOX11 reduced cartilage damage and lowered the OARSI score in mice, increased the hyaline cartilage layer thickness, reduced calcified cartilage thickness, inhibited ferroptosis and mitochondrial dysfunction, thereby alleviating OA.
There are some limitations here. Our results indicate that SOX11 reduces ferroptosis and mitochondrial dysfunction in chondrocytes subjected to IL-1β through enhancing transcription by directly binding to the promoter of NOX4, thereby alleviating OA. The core findings are mainly based on cell and animal models, and no systematic validation was conducted using human degenerated cartilage tissue samples. This leads to uncertainties regarding the expression profile and functional importance of the SOX11-NOX4 axis in the human OA pathological environment, which is a significant obstacle for clinical translation. Systematic validation of the expression profile of the SOX11-NOX4 axis in human degenerated cartilage tissue is crucial. Additionally, the means of knocking down SOX11 (local injection of viral vectors) might have issues such as insufficient targeting, long-term safety (such as off-target effects, immunogenicity), and administration methods (such as systemic delivery vs. local delivery), which are not in line with the actual needs of clinical treatment. At the same time, the research focuses on chondrocytes and does not assess the impact of SOX11 knockdown on other key cell types within the joint (such as synovial cells, osteoblasts, osteoclasts, and immune cells). In subsequent work, it is very important to elucidate the mechanism of the SOX11-NOX4 axis in regulating the production and release of SASP factors (such as IL1β, IL6, MMPs, etc.) in other key cells of OA. Moreover, in-depth research on the mechanism of the SOX11-NOX4 axis in regulating the production and release of SASP factors in chondrocytes and other cells within the joint is necessary. It is of great significance to further explore the potential mechanisms by which the SOX11-NOX4-ferroptosis-mitochondrial dysfunction axis mediated by ROS signaling [49], NF-κB signaling [50], Nrf2 signaling [51], or GRP78/GPX4 signaling [26] in OA (Supplementary Fig. S2).
Our research has revealed and systematically validated the pivotal role of the SOX11-NOX4-ferroptosis axis in the pathogenesis of OA. The core finding demonstrates that inhibiting SOX11 mitigates OA by alleviating mitochondrial dysfunction and ferroptosis in chondrocytes through reducing NOX4. This discovery establishes the novel “SOX11-NOX4-ferroptosis” signaling axis, which opens highly promising avenues for the development of targeted intervention strategies, diagnostic tools, and monitoring methods, thereby laying a critical theoretical foundation for achieving effective prevention, early diagnosis, and precision treatment of OA.
Acknowledgement: None.
Funding Statement: This work was sponsored by the Science Foundation of Hunan Aerospace Hospital (2025YJ13).
Author Contributions: Xingchang Fu and Gang Yang: Conceptualization, data curation, formal analysis, investigation, methodology, project administration, funding acquisition, resources, software, supervision, validation, visualization, writing—original draft, writing—review and editing. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: The data that support the findings of this study are available from the Corresponding Author, [Gang Yang], upon reasonable request.
Ethics Approval: The Animal Experiment Ethics Committee for Biomedical Research, Hunan Normal University approved all animal experiment procedures (No.: 2025-637). All experimental procedures were conducted in accordance with institutional guidelines for the use of experimental animals.
Conflicts of Interest: The authors declare no conflicts of interest.
Supplementary Materials: The supplementary material is available online at https://www.techscience.com/doi/10.32604/biocell.2026.074951/s1.
Abbreviations
| OA | Osteoarthritis |
| DMM | Destabilization of the medial meniscus |
| IL | Interleukin |
| SLC7A11 | Solute carrier family 7 member 11 |
| GPX4 | Glutathione peroxidase 4 |
| GSH | Glutathione |
| L-OOH | Lipid peroxides |
| ROS | Reactive oxygen species |
| ECM | Exacerbates extracellular matrix |
| SOX | Sex-determining region Y-box transcription factor |
| LPS | Lipopolysaccharide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NOX4 | NADPH oxidase 4 |
| SPF | Specific pathogen-free |
| H&E | Hematoxylin and eosin |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| ChIP | Chromatin immunoprecipitation |
| MDA | Malondialdehyde |
References
1. Tang SA, Zhang C, Oo WM, Fu K, Risberg MA, Bierma-Zeinstra SM, et al. Osteoarthritis. Nat Rev Dis Primers. 2025;11(1):10. doi:10.1038/s41572-025-00594-6. [Google Scholar] [PubMed] [CrossRef]
2. Zhu S, Qu W, He C. Evaluation and management of knee osteoarthritis. J Evid Based Med. 2024;17(3):675–87. doi:10.1111/jebm.12627. [Google Scholar] [PubMed] [CrossRef]
3. Yuan H, Yi N, Li D, Xu C, Yin GR, Zhuang C, et al. PPARγ regulates osteoarthritis chondrocytes apoptosis through caspase-3 dependent mitochondrial pathway. Sci Rep. 2024;14(1):11237. doi:10.1038/s41598-024-62116-w. [Google Scholar] [PubMed] [CrossRef]
4. Huang J, Chen Z, Wu Z, Xie X, Liu S, Kong W, et al. Geniposide stimulates autophagy by activating the GLP-1R/AMPK/mTOR signaling in osteoarthritis chondrocytes. Biomed Pharmacother. 2023;167(8):115595. doi:10.1016/j.biopha.2023.115595. [Google Scholar] [PubMed] [CrossRef]
5. Kang W, Xu Q, Dong H, Wang W, Huang G, Zhang J. Eriodictyol attenuates osteoarthritis progression through inhibiting inflammation via the PI3K/AKT/NF-κB signaling pathway. Sci Rep. 2024;14(1):18853. doi:10.1038/s41598-024-69028-9. [Google Scholar] [PubMed] [CrossRef]
6. Shawl M, Geetha T, Burnett D, Babu JR. Omega-3 supplementation and its effects on osteoarthritis. Nutrients. 2024;16(11):1650. doi:10.3390/nu16111650. [Google Scholar] [PubMed] [CrossRef]
7. Xia S, Li L, Shi Z, Sun N, He Y. Ferroptosis in osteoarthritis: metabolic reprogramming, immunometabolic crosstalk, and targeted intervention strategies. Front Immunol. 2025;16:1604652. doi:10.3389/fimmu.2025.1604652. [Google Scholar] [PubMed] [CrossRef]
8. Chen H, Han Z, Wang Y, Su J, Lin Y, Cheng X, et al. Targeting ferroptosis in bone-related diseases: facts and perspectives. J Inflamm Res. 2023;16:4661–77. doi:10.2147/JIR.S432111. [Google Scholar] [PubMed] [CrossRef]
9. Rochette L, Dogon G, Rigal E, Zeller M, Cottin Y, Vergely C. Lipid peroxidation and iron metabolism: two corner stones in the homeostasis control of ferroptosis. Int J Mol Sci. 2022;24(1):449. doi:10.3390/ijms24010449. [Google Scholar] [PubMed] [CrossRef]
10. Liu H, Deng Z, Yu B, Liu H, Yang Z, Zeng A, et al. Identification of SLC3A2 as a potential therapeutic target of osteoarthritis involved in ferroptosis by integrating bioinformatics, clinical factors and experiments. Cells. 2022;11(21):3430. doi:10.3390/cells11213430. [Google Scholar] [PubMed] [CrossRef]
11. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054–81. doi:10.1080/15548627.2020.1810918. [Google Scholar] [PubMed] [CrossRef]
12. Lu S, Liu Z, Qi M, Wang Y, Chang L, Bai X, et al. Ferroptosis and its role in osteoarthritis: mechanisms, biomarkers, and therapeutic perspectives. Front Cell Dev Biol. 2024;12:1510390. doi:10.3389/fcell.2024.1510390. [Google Scholar] [PubMed] [CrossRef]
13. Zhou X, Pan Y, Li J, Zhuang R, Tong P, Xia H. Notopterol mitigates osteoarthritis progression and relieves pain in mice by inhibiting PI3K/Akt/GPX4-mediated ferroptosis. Int Immunopharmacol. 2025;151(1):114323. doi:10.1016/j.intimp.2025.114323. [Google Scholar] [PubMed] [CrossRef]
14. Kazmirczak F, Vogel NT, Prisco SZ, Patterson MT, Annis J, Moon RT, et al. Ferroptosis-mediated inflammation promotes pulmonary hypertension. Circ Res. 2024;135(11):1067–83. doi:10.1161/CIRCRESAHA.123.324138. [Google Scholar] [PubMed] [CrossRef]
15. Xu Y, Gu X, Li X, Chen Y, Wei Z, Wang J, et al. β-diketone functionalized microspheres chelate reactive iron via metal coordination for cartilage repair. Adv Healthc Mater. 2025;14(10):2403933. doi:10.1002/adhm.202403933. [Google Scholar] [PubMed] [CrossRef]
16. Sheng W, Liao S, Wang D, Liu P, Zeng H. The role of ferroptosis in osteoarthritis: progress and prospects. Biochem Biophys Res Commun. 2024;733:150683. doi:10.1016/j.bbrc.2024.150683. [Google Scholar] [PubMed] [CrossRef]
17. Ahmed EA, Alzahrani AM. SOXC transcription factors as diagnostic biomarkers and therapeutic targets for arthritis. Int J Mol Sci. 2023;24(4):4215. doi:10.3390/ijms24044215. [Google Scholar] [PubMed] [CrossRef]
18. Bhattaram P, Muschler G, Wixler V, Lefebvre V. Inflammatory cytokines stabilize SOXC transcription factors to mediate the transformation of fibroblast-like synoviocytes in arthritic disease. Arthritis Rheumatol. 2018;70(3):371–82. doi:10.1002/art.40386. [Google Scholar] [PubMed] [CrossRef]
19. Xu F, Hu QF, Li J, Shi CJ, Luo JW, Tian WC, et al. SOX4-activated lncRNA MCM3AP-AS1 aggravates osteoarthritis progression by modulating miR-149-5p/Notch1 signaling. Cytokine. 2022;152:155805. doi:10.1016/j.cyto.2022.155805. [Google Scholar] [PubMed] [CrossRef]
20. Xu S, Yu J, Wang Z, Ni C, Xia L, Tang T. SOX11 promotes osteoarthritis through induction of TNF-α. Pathol Res Pract. 2019;215(7):152442. doi:10.1016/j.prp.2019.152442. [Google Scholar] [PubMed] [CrossRef]
21. Pan W, Wang H, Ruan J, Zheng W, Chen F, Kong J, et al. lncRNA myocardial infarction-associated transcript (MIAT) knockdown alleviates LPS-induced chondrocytes inflammatory injury via regulating miR-488-3p/sex determining region Y-related HMG-box 11 (SOX11) axis. Open Life Sci. 2021;16(1):511–22. doi:10.1515/biol-2021-0023. [Google Scholar] [PubMed] [CrossRef]
22. Wang X, Fan J, Ding X, Sun Y, Cui Z, Liu W. Tanshinone I inhibits IL-1β-induced apoptosis, inflammation and extracellular matrix degradation in chondrocytes CHON-001 cells and attenuates murine osteoarthritis. Drug Des Devel Ther. 2019;13:3559–68. doi:10.2147/DDDT.S216596. [Google Scholar] [PubMed] [CrossRef]
23. Chen H, Ao Q, Wang Y, Qian Y, Cheng Q, Zhang W. SOX11 as a potential prognostic biomarker in hepatocellular carcinoma linked to immune infiltration and ferroptosis. Chin J Cancer Res. 2024;36(4):378–97. doi:10.21147/j.issn.1000-9604.2024.04.03. [Google Scholar] [PubMed] [CrossRef]
24. Li J, Wang L, Wang B, Zhang Z, Jiang L, Qin Z, et al. NOX4 is a potential therapeutic target in septic acute kidney injury by inhibiting mitochondrial dysfunction and inflammation. Theranostics. 2023;13(9):2863–78. doi:10.7150/thno.81240. [Google Scholar] [PubMed] [CrossRef]
25. Park MW, Cha HW, Kim J, Kim JH, Yang H, Yoon S, et al. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. 2021;41:101947. doi:10.1016/j.redox.2021.101947. [Google Scholar] [PubMed] [CrossRef]
26. Wang Q, Qi B, Shi S, Jiang W, Li D, Jiang X, et al. Melatonin alleviates osteoarthritis by regulating NADPH oxidase 4-induced ferroptosis and mitigating mitochondrial dysfunction. J Pineal Res. 2024;76(6):e12992. doi:10.1111/jpi.12992. [Google Scholar] [PubMed] [CrossRef]
27. Charan J, Kantharia ND. How to calculate sample size in animal studies? J Pharmacol Pharmacother. 2013;4(4):303–6. doi:10.4103/0976-500X.119726. [Google Scholar] [PubMed] [CrossRef]
28. Zhang S, Zhang B, Liao Z, Chen Y, Guo W, Wu J, et al. Hnrnpk protects against osteoarthritis through targeting WWC1 mRNA and inhibiting Hippo signaling pathway. Mol Ther. 2024;32(5):1461–78. doi:10.1016/j.ymthe.2024.02.027. [Google Scholar] [PubMed] [CrossRef]
29. Zhou X, Zheng Y, Sun W, Zhang Z, Liu J, Yang W, et al. D-mannose alleviates osteoarthritis progression by inhibiting chondrocyte ferroptosis in a HIF-2α-dependent manner. Cell Prolif. 2021;54(11):e13134. doi:10.1111/cpr.13134. [Google Scholar] [PubMed] [CrossRef]
30. Afzal M, Rekha MM, Sahoo S, Pandey SN, Maji C, Goyal K, et al. Targeting the senescence-associated secretory phenotype to modify osteoarthritis in aging. Inflammopharmacology. 2025;33(11):6555–75. doi:10.1007/s10787-025-02001-8. [Google Scholar] [PubMed] [CrossRef]
31. Gong Z, Zhu J, Chen J, Feng F, Zhang H, Zhang Z, et al. CircRREB1 mediates lipid metabolism related senescent phenotypes in chondrocytes through FASN post-translational modifications. Nat Commun. 2023;14(1):5242. doi:10.1038/s41467-023-40975-7. [Google Scholar] [PubMed] [CrossRef]
32. Yao X, Sun K, Yu S, Luo J, Guo J, Lin J, et al. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J Orthop Translat. 2020;27:33–43. doi:10.1016/j.jot.2020.09.006. [Google Scholar] [PubMed] [CrossRef]
33. Chan WCW, Tan Z, To MKT, Chan D. Regulation and role of transcription factors in osteogenesis. Int J Mol Sci. 2021;22(11):5445. doi:10.3390/ijms22115445. [Google Scholar] [PubMed] [CrossRef]
34. Nishimura R, Hata K, Takahata Y, Murakami T, Nakamura E, Ohkawa M, et al. Role of signal transduction pathways and transcription factors in cartilage and joint diseases. Int J Mol Sci. 2020;21(4):1340. doi:10.3390/ijms21041340. [Google Scholar] [PubMed] [CrossRef]
35. Huang Y, Wang Z. Therapeutic potential of SOX family transcription factors in osteoarthritis. Ann Med. 2025;57(1):2457520. doi:10.1080/07853890.2025.2457520. [Google Scholar] [PubMed] [CrossRef]
36. Jing X, Du T, Li T, Yang X, Wang G, Liu X, et al. The detrimental effect of iron on OA chondrocytes: importance of pro-inflammatory cytokines induced iron influx and oxidative stress. J Cell Mol Med. 2021;25(12):5671–80. doi:10.1111/jcmm.16581. [Google Scholar] [PubMed] [CrossRef]
37. Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 2020;30(10):3411–23.e7. doi:10.1016/j.celrep.2020.02.049. [Google Scholar] [PubMed] [CrossRef]
38. He Q, Yang J, Pan Z, Zhang G, Chen B, Li S, et al. Biochanin A protects against iron overload associated knee osteoarthritis via regulating iron levels and NRF2/System xc-/GPX4 axis. Biomed Pharmacother. 2023;157:113915. doi:10.1016/j.biopha.2022.113915. [Google Scholar] [PubMed] [CrossRef]
39. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22(2):225–34. doi:10.1038/s41556-020-0461-8. [Google Scholar] [PubMed] [CrossRef]
40. Cao S, Wei Y, Xiong A, Yue Y, Yang J, Wang D, et al. Paeonol inhibits ACSL4 to protect chondrocytes from ferroptosis and ameliorates osteoarthritis progression. J Orthop Transl. 2025;50:1–13. doi:10.1016/j.jot.2024.10.005. [Google Scholar] [PubMed] [CrossRef]
41. Xu Y, Yang Z, Dai T, Xue X, Xia D, Feng Z, et al. Characteristics and time points to inhibit ferroptosis in human osteoarthritis. Sci Rep. 2023;13(1):21592. doi:10.1038/s41598-023-49089-y. [Google Scholar] [PubMed] [CrossRef]
42. Bonanni R, Falvino A, Smakaj A, Tranquillo L, Rinaldi AM, D’Arcangelo G, et al. Trolox, r-irisin and resveratrol cocktail to counteract osteoblast metabolism alterations in osteoarthritis and osteoporosis. J Bone Miner Metab. 2025;43(6):672–84. doi:10.1007/s00774-025-01642-7. [Google Scholar] [PubMed] [CrossRef]
43. Coryell PR, Diekman BO, Loeser RF. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat Rev Rheumatol. 2021;17(1):47–57. doi:10.1038/s41584-020-00533-7. [Google Scholar] [PubMed] [CrossRef]
44. Jiang N, Xing B, Peng R, Shang J, Wu B, Xiao P, et al. Inhibition of Cpt1a alleviates oxidative stress-induced chondrocyte senescence via regulating mitochondrial dysfunction and activating mitophagy. Mech Ageing Dev. 2022;205:111688. doi:10.1016/j.mad.2022.111688. [Google Scholar] [PubMed] [CrossRef]
45. Wei W, Yang L, Wang B, Tang L, Li J, Liu C, et al. Remote ischemic preconditioning attenuates mitochondrial dysfunction and ferroptosis of tubular epithelial cells by inhibiting NOX4-ROS signaling in acute kidney injury. Int J Biol Sci. 2025;21(5):2313–29. doi:10.7150/ijbs.105667. [Google Scholar] [PubMed] [CrossRef]
46. Wang S, Li W, Zhang P, Wang Z, Ma X, Liu C, et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. J Adv Res. 2022;41:63–75. doi:10.1016/j.jare.2022.01.004. [Google Scholar] [PubMed] [CrossRef]
47. Chen J, Deng X, Lin T, Huang J, Yang Y, Lian N. Ferrostatin-1 reversed chronic intermittent hypoxia-induced ferroptosis in aortic endothelial cells via reprogramming mitochondrial function. Nat Sci Sleep. 2024;16:401–11. doi:10.2147/NSS.S442186. [Google Scholar] [PubMed] [CrossRef]
48. Wang X, Liu Z, Peng P, Gong Z, Huang J, Peng H. Astaxanthin attenuates osteoarthritis progression via inhibiting ferroptosis and regulating mitochondrial function in chondrocytes. Chem Biol Interact. 2022;366:110148. doi:10.1016/j.cbi.2022.110148. [Google Scholar] [PubMed] [CrossRef]
49. Chen H, Nie P, Li J, Wu Y, Yao B, Yang Y, et al. Cyclophosphamide induces ovarian granulosa cell ferroptosis via a mechanism associated with HO-1 and ROS-mediated mitochondrial dysfunction. J Ovarian Res. 2024;17(1):107. doi:10.1186/s13048-024-01434-z. [Google Scholar] [PubMed] [CrossRef]
50. Huang J, Meng P, Liang Y, Li X, Zhou S, Li J, et al. Tubular CD44 plays a key role in aggravating AKI through NF-κB p65-mediated mitochondrial dysfunction. Cell Death Dis. 2025;16(1):119. doi:10.1038/s41419-025-07438-x. [Google Scholar] [PubMed] [CrossRef]
51. Cao G, Yang C, Jin Z, Wei H, Xin C, Zheng C, et al. FNDC5/irisin reduces ferroptosis and improves mitochondrial dysfunction in hypoxic cardiomyocytes by Nrf2/HO-1 axis. Cell Biol Int. 2022;46(5):723–36. doi:10.1002/cbin.11763. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools