
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Obesity, Metabolic Dysfunction-Associated Steatotic Liver Disease and Hepatocellular Carcinoma: How Molecular Changes Impact Cellular Functions
1 Institute of Molecular Pathobiochemistry, Experimental Gene Therapy, and Clinical Chemistry (IFMPEGKC), RWTH University Hospital Aachen, Pauwelsstr. 30, Aachen, D-52074, Germany
2 AOU Modena, Department of Internal Medicine, Ospedale Civile di Baggiovara (-2023), Modena, Italy
* Corresponding Author: Amedeo Lonardo. Email:
(This article belongs to the Special Issue: Molecular Insights into the Obesity-Cancer Nexus: From Cellular Mechanisms to Therapeutic Targets)
BIOCELL 2026, 50(5), 1 https://doi.org/10.32604/biocell.2026.076177
Received 15 November 2025; Accepted 19 January 2026; Issue published 13 May 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
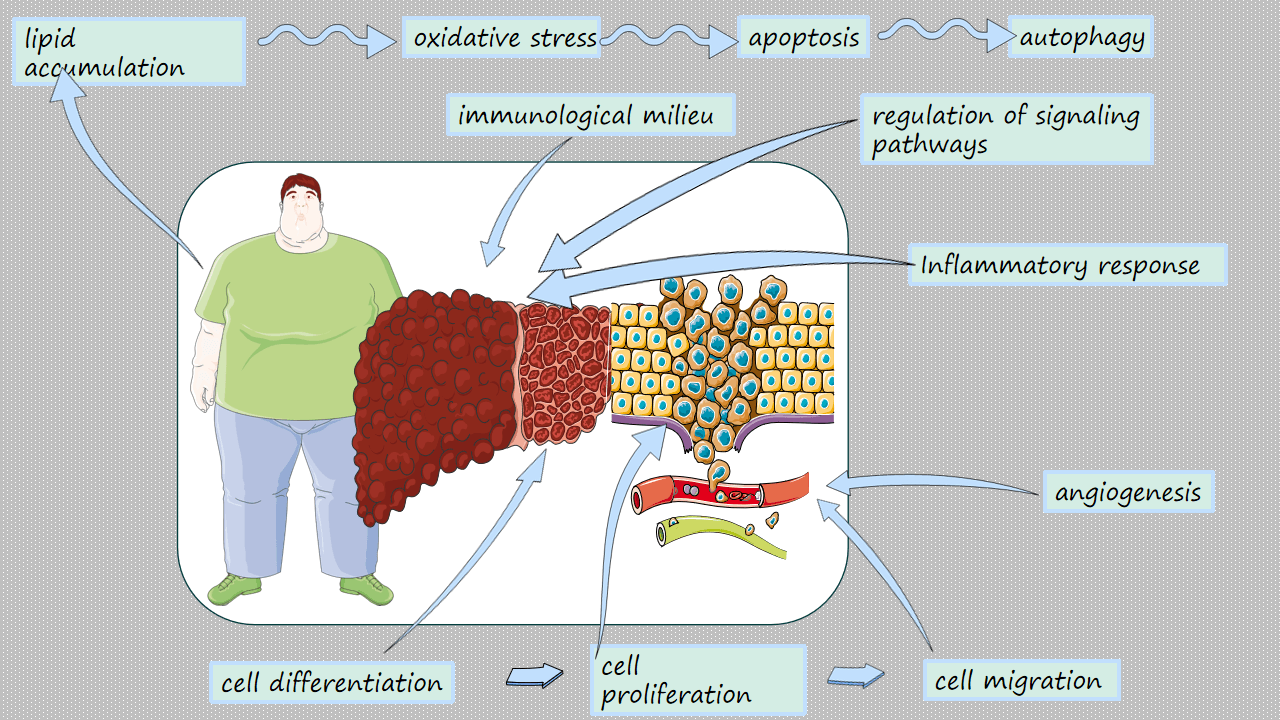
Obesity is a complex chronic condition characterized by an excess of body fat that manifests in various clinical pathophenotypes, each affecting liver health differently. One significant cause of chronic liver diseases among those living with obesity is metabolic dysfunction-associated steatotic liver disease (MASLD), which is linked to one or more cardiometabolic risk factors in individuals who do not engage in harmful alcohol consumption. Hepatocellular carcinoma (HCC) is the most common form of primary liver cancer and is increasingly being associated with MASLD through intricate immunological, cellular, proinflammatory, molecular, and genetic mechanisms. In this review, we examine the molecular changes and altered functions of hepatocytes related to lipid accumulation, oxidative stress, inflammatory responses, and the regulation of signaling pathways such as mechanistic target of rapamycin (mTOR), AMP-activated protein kinase (AMPK), and nuclear factor kappa B (NF-κB). We also explore how these processes impact cell proliferation, apoptosis, autophagy, migration, the immunological environment, angiogenesis, and cell differentiation. Finally, we discuss the challenges posed by MASLD-HCC and how these may be effectively managed by promoting effective risk stratification, early diagnosis, and innovative treatment schedules for this numerically growing category of patients.Graphic Abstract
Keywords
1 Background, Definitions, and Aims
Obesity is a complex chronic condition characterized by the excessive accumulation of adipose tissue, presenting significant challenges to both individual health and public health systems globally [1]. This disease encompasses various pathophenotypes, such as android, gynoid, sarcopenic, metabolically healthy, and unhealthy obesity, each with different associations with liver health in the spectrum of chronic liver disease (CLD). This spectrum includes alcohol-related CLD, viral hepatitis B and C, and metabolic dysfunction-associated steatotic liver disease (MASLD), which is considered the primary manifestation of obesity-related CLD [2]. Body mass index (BMI) has its limitations, as it may misclassify obesity in individuals with well-developed muscle mass or fluid retention [3]. Visceral adiposity is a better indicator of metabolic dysfunction than BMI, making waist circumference a useful sex- and ethnic-specific anthropometric measurement [4]. Recognizing the constraints of BMI, experts have introduced the concept of “clinical obesity”, which identifies a chronic, systemic illness characterized by excess adiposity leading to impaired bodily functions [5].
1.2 Metabolic Dysfunction-Associated Steatotic Liver Disease
MASLD encompasses simple steatosis, metabolic dysfunction-associated steatohepatitis [MASH, previously known as nonalcoholic steatohepatitis (NASH)], fibrosis, cirrhosis, and MASLD-hepatocellular carcinoma (HCC). MASLD is the type of steatotic liver disease (SLD) associated with one or more cardiometabolic risk factors in individuals who do not consume excessive alcohol [6]. MASLD is the most recent term used to describe the accumulation of fatty substances in the liver along with systemic metabolic dysfunction, replacing previous terms like nonalcoholic fatty liver disease (NAFLD) and metabolic dysfunction-associated fatty liver disease (MAFLD) [7].
HCC, the most common histological subtype of primary liver cancer (PLC), develops in the context of CLD from various causes. It has a high mortality rate, and its epidemiological, clinical, and financial burden is expected to increase [8]. While viral CLD has declined in recent years, MASLD has been on the rise, with metabolic comorbidities identified as risk factors and co-factors for HCC through complex genetic, immunological, cellular, proinflammatory, and molecular processes [9]. Understanding these mechanisms necessitates close surveillance and early diagnosis of at-risk patients to pave the way for personalized medicine, chemoprevention, and innovative management of MASLD-HCC [10].
Given the rapidly evolving clinical and epidemiological landscape, our aim is to highlight how molecular changes impact cellular functions in the context of obesity-related MASLD-HCC.
2 Criteria of Bibliographic Research
This narrative review explores the molecular mechanisms connecting obesity with MASLD and HCC. To set up this article, we conducted research on PubMed/MEDLINE and the Web of Science Core Collection for relevant articles. In our search strategy we combined Medical Subject Headings and free-text terms related to “obesity”, “body mass index”, “adiposity”, “MASLD”, “MAFLD”, “NAFL”, “NAFLD”, “MASH”, “NASH”, “hepatocellular carcinoma”, “liver cancer”, “pathogenesis”, “immunometabolism”, “oxidative stress”, “mTOR”, “AMPK”, “NF-κB”, “angiogenesis”, “diagnosis”, “surveillance”, and “therapy”. We focused on studies that reported findings in humans and studies published in the English language. The identified sources included peer-reviewed original investigations, systematic reviews, meta-analyses, clinical guidelines, and high-impact narrative reviews that addressed epidemiology, molecular mechanisms, risk stratification, diagnostics, or therapeutic interventions relevant to the obesity/MASLD/HCC axis.
3 Burden and Risk Factors for Obesity, MASLD, and HCC
Affecting more than 1 billion individuals, approximately 13% of the population as of 2022, obesity represents a significant global health concern. The prevalence of obesity has tripled since 1975 and is projected to impact 1.9 billion adults (25%) by 2035 and 3.8 billion, surpassing half of the worldwide adult population, by 2050 [11]. Historically more prevalent in high-income nations, obesity rates are now increasing most rapidly in low- and middle-income nations, with over 70% of obese individuals living in developing countries [12]. This escalating burden contributes to over 5 million annual deaths related to conditions such as type 2 diabetes, cardiovascular diseases, and various cancers, notably including HCC [11,13]. Consequently, the economic impact of obesity is substantial, with estimates indicating that it may account for more than 3% of global Gross Domestic Product expenditures worldwide by 2060 [14]. As for the causes, obesity involves multiple interactions between genetic, hormonal, behavioral, environmental, and socio-economic determinants, with urbanization dynamics and globalization attitudes facilitating sedentary lifestyles and promoting consumption of calorie-dense, ultra-processed, and insecure foodstuffs [11,15,16].
3.2 Metabolic Dysfunction-Associated Steatotic Liver Disease
An accurate understanding of MASLD epidemiology is essential for planning healthcare resource allocation and health policy decisions [17]. The prevalence of MASLD, MASLD-cirrhosis, MASLD-related end-stage liver disease awaiting liver transplantation, and MASLD-HCC is increasing globally, driven by factors such as an aging general population and rising rates of diabesity [17]. Using data from the 2017 to 2023 edition of the National Health and Nutrition Examination Survey (NHANES), Paik et al. [18] found that the age-standardized prevalence of MASLD was 32.42% with clear sex disparities and ethnic gradients: men had a significantly higher prevalence than women, and Mexicans had the highest burden, followed by Whites, Hispanics, Asians, and Blacks. A meta-analytic review of 35 published studies totaling 513,742 individuals [19] found that the pooled global prevalence of cirrhosis among patients with MASLD was estimated at 3.26% (95% CI: 2.47%–4.31%) in general practice settings and 14.51% (95% CI: 11.22%–18.57%) among individuals in inpatient settings or those referred to liver biopsy. Regionally, higher prevalence rates in high-risk settings were observed in North America and Australia (18.38%; 95% CI: 9.06%–33.75%), followed by Europe (10.16%; 95% CI: 5.71%–17.44%) and Asia (9.12%; 95% CI: 6.11%–13.40%) (p = 0.007). Additionally, diagnoses based on ICD coding indicated a significantly greater prevalence of cirrhosis (27.43%) compared to cases confirmed by liver biopsy (13.24%; p < 0.001). The estimated pooled all-cause mortality rate for patients with MASLD-related cirrhosis was 7.91 per 100 person-years (95% CI: 4.44–13.71), as reported across nine studies [19].
The age-standardized incidence rate (ASIR) increased from 475.54 to 593.28 per 100,000 (AAPC 0.71). MASLD was responsible for 138,328 deaths, with a higher mortality rate observed among females (52.18%). The regions of East Asia, South Asia, and North Africa/Middle East reported the highest prevalence and incidence rates, while Western Europe experienced the most rapid growth. Projections indicate that by 2045, the ASIR will reach 928.10 per 100,000, resulting in an estimated 667.58 million new cases, predominantly affecting males [20]. Various explanations may clarify the reasons behind the significant differences in cirrhosis prevalence estimates in published studies [21]. Patient selection varies by study type, such as population-based samples, referral centers, or patients with advanced disease or comorbidities. Diagnostic approaches also differ, ranging from non-invasive scores and imaging to liver biopsy, with varying cirrhosis cut-offs. Additionally, geographical and healthcare system differences can introduce selection bias that affects referrals to specialized care.
Without interventions, the clinical, economic, and quality-of-life burden of MASH from 2021 to 2040 is projected to increase across the United States, Western Europe, the United Kingdom, Japan, Brazil, and Saudi Arabia, highlighting the urgent need for both national and global strategies to reduce the negative impact of MASH on individuals and society [22]. By very definition, MASLD is intricately linked with systemic metabolic dysfunction, and therefore this disorder progresses in parallel with the individual features of the metabolic syndrome, comprising, further to obesity, altered glucose metabolism, atherogenic dyslipidemia, and arterial hypertension. In MASLD, the risks of fibrotic progression and MASLD-related mortality are directly proportional to the number of individual components of the metabolic syndrome [23].
Wu et al. reported that in 2021, PLC attributed to elevated BMI and/or high fasting plasma glucose (FPG) resulted in 59,970 deaths globally and accounted for 1,540,437 disability-adjusted life years [13]. Additionally, from 1990 to 2021, there has been a consistent global increase in liver cancer due to metabolic risks, with high BMI identified as the predominant risk factor. The highest mortality and Disability-Adjusted Life Year (DALY) burdens were observed in countries with high Socio-Demographic Index (SDI), while the most rapid increase occurred in low-middle SDI countries. The burdens associated with elevated BMI and FPG were greater among men compared to women [13]. Another study has indicated reductions in PLC related to chronic viral hepatitis, contrasted with increased rates of PLC associated with MASLD, particularly in high-SDI regions and among those aged over 75 [24]. There is a global male-to-female ratio of 2.19, though sex differences vary on a regional basis. Finally, the incidence rate is projected to persistently rise from 2021 to 2050 [24].
Elevated FPG indicates systemic insulin resistance and hyperinsulinemia, which activate insulin/IGF-1 pathways. This activation promotes cell proliferation, inhibits apoptosis, and increases oxidative stress and DNA damage in hepatocytes [25]. It should be emphasized that the BMI/FPG attributable fraction of PLC predominantly influences MASLD and MASLD-cirrhosis as intermediate phenotypes [13].
4 Molecular and Cellular Pathogenesis of Obesity-Associated Hepatocellular Carcinoma
The earliest morphological feature of MASLD is lipid accumulation in hepatocytes, which represents the biochemical basis for progressive liver injury and hepatocarcinogenesis. In MASLD, excessive storage of triglycerides is closely linked to disruptions in systemic energy balance related to obesity and concurrent cardiometabolic risk factors, independent of harmful alcohol consumption. Moreover, hepatocyte steatosis triggers oxidative stress, inflammatory responses, and abnormal signaling through nutrient-sensing pathways such as mechanistic target of rapamycin complex 1 (mTORC1), AMP-activated protein kinase (AMPK), and nuclear factor kappa B (NF-κB) [26,27,28]. Mechanistically, cellular hepatic lipid overload originates from three main sources: increased delivery of dietary fatty acids and chylomicron remnants, increased adipose tissue lipolysis releasing non-esterified fatty acids into the portal circulation, and augmented de novo lipogenesis (DNL) due to hyperinsulinemia and glucose influx (Fig. 1) [29].

Figure 1: Hepatic lipid overload and its consequences in obesity-related MASLD. Three major sources of hepatic lipid influx, including dietary fatty acids and chylomicron remnants, adipose tissue lipolysis due to insulin resistance, and increased de novo lipogenesis driven by SREBP-1c and ChREBP, converge to promote triglyceride-rich lipid droplet accumulation in hepatocytes. This process is modulated by genetic variants in genes encoding PNPLA3, TM6SF2, MBOAT7, and GCKR. Excess saturated fatty acids and lipotoxic intermediates induce endoplasmic reticulum (ER) stress, mitochondrial and peroxisomal reactive oxygen species (ROS) production, and oxidative damage. These factors, combined with damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), and cytokine release, fuel inflammation, activate hepatic stellate cells (HSCs), and drive fibrogenesis, portal hypertension, and might lead to cirrhosis and hepatocellular carcinoma (HCC). This lowers the threshold for hepatocarcinogenesis. This original illustration was created using Servier Medical ART (SMART) and is licensed under the Creative Commons Attribution 4.0 International License (CC BY 4.0).
Insulin resistance, a hallmark of obesity, impairs the antilipolytic effect of insulin in peripheral adipose tissue while enhancing its lipogenic effect in the liver through sterol regulatory element-binding protein-1c (SREBP-1c) [30]. Additionally, carbohydrate-responsive element-binding protein (ChREBP), which responds to fructose and glucose signals, has been considered a driving factor in DNL [30]. The excess of saturated long-chain fatty acids like palmitate is converted to triglycerides and stored in dynamic, endoplasmic reticulum (ER)-derived lipid droplets, initially considered harmless but now recognized as lipotoxic when protective mechanisms are overwhelmed [31].
While the majority of MASLD cases still arise from environmental or metabolic drivers in genetically “average” individuals, genetic factors also play a key pathogenic role in MASLD. In humans, gene variants such as PNPLA3 I148M, TM6SF2 E167K, MBOAT7 rs641738, and Glucokinase Regulatory Protein (GCKR) highlight the importance of lipid droplet biology, VLDL secretion, and phospholipid remodeling in MASLD [32].
The PNPLA3 I148M gene variant in humans reduces lipase activity at the lipid droplet surface, leading to the retention of triglyceride-rich droplets enriched in polyunsaturated fatty acids and promotes steatogenesis through the development of macrovesicular steatosis [33]. These droplets are more susceptible to lipid peroxidation, which amplifies oxidative and inflammatory signaling.
TM6SF2 E167K impairs VLDL secretion, resulting in intrahepatic triglyceride accumulation and altered lipid composition that enhances lipotoxic stress despite lower circulating lipids [34]. Unlike typical cases of obese or metabolic MASLD, individuals with this variant do not present with hypertriglyceridemia [35,36] and have a reduced risk of cardiovascular disease, supporting the notion that “not all fat is alike in MASLD” [37,38].
MBOAT7 rs641738 disrupts phosphatidylinositol remodeling, shifting membrane phospholipid composition toward arachidonic-acid-poor species. This influences membrane fluidity, receptor clustering, and the generation of pro-inflammatory eicosanoids [39,40].
The GCKR gene is also implicated in susceptibility to MASLD. The rs1260326 C > T variant enhances hepatic glucose uptake and de novo lipogenesis, resulting in increased storage of intrahepatic lipids [32].
Several of these polymorphisms not only increase the severity of steatosis but also enhance oxidative stress and fibrogenic signaling, lowering the threshold for carcinogenesis. At the cellular level, excess saturated fatty acids alter membrane composition, induce ER stress, and activate unfolded protein response pathways mediated by the eIF2 kinase PERKα, ATF6, and IRE1 [41,42]. This in turn triggers c-Jun N-terminal kinase (JNK) and nuclear factor-κB (NF-κB) cascades. Reactive oxygen species (ROS) from overloaded mitochondrial and peroxisomal oxidation damage DNA, disrupt electron transport chain integrity, and initiate lipid peroxidation, amplifying mutagenic and pro-oncogenic signals [42].
Moreover, hepatic steatosis disrupts the sinusoidal microenvironment at the organ level. In particular, lipid-laden hepatocytes release danger-associated molecular patterns (DAMPs) and several metabolites like ceramides and diacylglycerols, which recruit and activate immune cells like Kupffer cells and macrophages [43]. These cells produce inflammatory cytokines, including tumor-necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), promoting insulin resistance, enhancing lipogenesis, and creating a pro-inflammatory environment, setting the soil for cancer development [35]. Simultaneously, lipotoxic intermediates activate hepatic stellate cells, promoting their transdifferentiation to contractile myofibroblasts, thereby driving liver fibrosis that hinders nutrient and oxygen diffusion, maintaining hypoxia-inducible factor-1α (HIF-1α) and facilitating angiogenesis, creating favorable conditions for the development of portal hypertension, and the proliferation of dysplastic hepatocyte nodules [36].
Nutrient-sensing pathways are closely linked to steatosis. Excess caloric intake activates the mechanistic target of rapamycin complex 1 (mTORC1), promoting DNL and inhibiting autophagy, including lipophagy, which, in turn, limits the ability of the liver to mobilize stored triglycerides [44]. Conversely, in MASLD, increased AMP-activated protein kinase (AMPK) activity can inhibit the synthesis of fatty acids and cholesterol by downregulating the expression of genes involved in steatogenesis, such as fatty acid synthase, SREBP-1c, acetyl-CoA carboxylase, and HMG-CoA reductase [45]. Therapeutic interventions that restore AMPK activity, such as the administration of metformin or increasing physical activity, or inhibition of the mTORC1 pathway by rapamycin analogs (i.e., rapalogs), have been shown to reverse steatosis and reduce HCC risk in preclinical models [46].
The distribution of lipid droplets also impacts oncogenic signaling. Particularly, macrosteatosis, characterized by large, macrovesicular cellular fat droplets, can displace the nucleus, provoking gene expression differences, while microvesicular steatosis is associated with mitochondrial dysfunction and increased ROS [47,48,49]. Advanced imaging and single-cell transcriptomics have revealed diversity in lipid droplet size and properties, suggesting that specific lipid signatures may confer carcinogenic potential. Therefore, lipid accumulation in MASLD is not just a simple, passive storage process but an active driver of hepatocyte damage, fibrosis, portal hypertension, and tumorigenesis through interconnected metabolic, oxidative, inflammatory, and signaling pathways.
The transition from simple steatosis in MASLD to progressive liver injury that can ultimately lead to HCC is critically driven by sustained oxidative stress, which acts in concert with lipid accumulation, inflammation, and dysregulated signaling pathways to damage hepatocytes and remodel the hepatic microenvironment [50]. As highlighted above, steatotic hepatocytes experience heightened ROS generation that can contribute to mutagenesis, fibrogenesis, and oncogenesis, thereby forming a central pathobiological link between obesity, MASLD, and HCC.
Sources of ROS generation in MASLD are multifactorial. Caloric excess increases the flux of free fatty acids and fructose into hepatocytes, leading to excessive β-oxidation and tricarboxylic acid cycle overload. This saturates the mitochondrial electron transport chain, leading to abnormally high concentrations of nicotinamide adenine dinucleotide (NADH) and nicotinamide adenine dinucleotide phosphate (NADPH) that create a “traffic jam”, causing a partial reversal and leakage of the electron flow at complexes I and III with subsequent formation of superoxide anions and hydrogen peroxide [51]. Apart from ROS generation in mitochondria, enzymes such as NADPH oxidase (NOX), xanthine oxidase (XO), cytochrome P450 2E1 (CYP2E1), cyclooxygenases, and lipoxygenases in the cytosol and plasma membranes also produce ROS, especially hydrogen peroxide [52]. In parallel, increased ER stress triggered by lipoprotein synthesis and by lipid-induced membrane perturbation activates the unfolded protein response (UPR), which elevates ROS through disulfide-bond formation and calcium leakage from the ER, thereby further leading to disease progression by progressing to cell death and dynamic pathophysiological changes [53]. Together, these “multiple hits” overwhelm cellular antioxidant defense mechanisms, shifting the redox balance toward sustained oxidative injury.
Beyond mitochondria, fatty acid-induced oxidative stress in MASLD is also fueled by peroxisomes and by the oxidative protein-folding machinery of the ER [54]. In steatotic hepatocytes, very-long-chain and branched-chain fatty acids that cannot be efficiently oxidized in mitochondria are shunted to peroxisomes. There, acyl-CoA oxidases catalyze the first step of β-oxidation and transfer electrons directly to molecular oxygen, generating stoichiometric amounts of hydrogen peroxide. Under chronic lipotoxic conditions, this peroxisomal flux can exceed the detoxifying capacity of catalase, allowing hydrogen peroxide to diffuse into the cytosol and neighboring organelles. This amplifies redox stress and sustains lipid peroxidation. In parallel, the ER lumen is intrinsically oxidizing to permit correct disulfide-bond formation during protein folding. Thiol-disulfide exchange reactions mediated by protein disulfide isomerase and ER oxidoreductin 1 (Ero1) couple oxidative folding of nascent secretory and membrane proteins to the reduction of oxygen, producing hydrogen peroxide as a by-product [55]. The increased demand for lipoprotein assembly, cytokine and hormone receptors, and acute-phase proteins in MASLD augments this oxidative folding load [56,57]. This, together with lipid-induced membrane perturbation, promotes the accumulation of misfolded proteins, activation of the unfolded protein response, and further reactive species generation. Thus, peroxisomal β-oxidation and ER oxidative protein folding cooperate with mitochondrial dysfunction to maintain a self-perpetuating cycle of oxidative stress that drives hepatocyte injury, fibrogenesis, and, eventually, hepatocarcinogenesis [56].
At the molecular level, ROS induces direct damage to lipids, proteins, and nucleic acids [47]. Lipid peroxidation generates highly reactive aldehydes such as 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), which form adducts with DNA and histones, fostering mutagenic lesions that predispose to malignant transformation [58,59]. Oxidative DNA damage, exemplified by 8-oxo-2′-deoxyguanosine formation, representing the most common form of oxidatively damaged DNA, TP53 and telomerase reverse transcriptase gene mutations, genomic instability, and chromosomal rearrangements, are hallmarks of HCC [60,61]. At the same time, ROS oxidize protein thiols, disrupting enzymatic activity and favoring protein misfolding, which further perpetuates ER stress and activates JNK, NF-κB, and mitogen-activated protein kinase (MAPK) pathways [62]. These signaling cascades promote hepatocyte apoptosis or, paradoxically, survival and proliferation in precancerous clones, depending on the redox threshold and cellular context. Importantly, ROS themselves are harmful epigenetic regulators that interfere with DNA methylation, histone-modifying enzymes, and RNA modification, accelerating epigenomic re-programming toward oncogenic phenotypes, serving as an exemplar of “mutagenic oxidative stress” [63].
Oxidative stress also orchestrates crosstalk between parenchymal and non-parenchymal cells. Damaged hepatocytes release DAMPs such as mitochondrial DNA and oxidized phospholipids, which engage toll-like receptors (TLRs) on Kupffer cells and recruit infiltrating monocyte-derived macrophages [64]. The resulting production of TNF-α, IL-1β, and IL-6 amplifies ROS generation through NOXs, establishing a vicious circle of oxidative injury and inflammation. In parallel, hydrogen peroxide and 4-HNE activate hepatic stellate cells, triggering the transition to a myofibroblast cell phenotype (i.e., the myofibroblast), capable of producing and secreting large quantities of extracellular matrix compounds, triggering hepatic fibrosis [58,65]. The fibrotic milieu impairs sinusoidal perfusion, resulting in portal hypertension, and exacerbates local hypoxia. It induces HIF-1α, which further stimulates angiogenesis and glycolytic re-programming, additional factors driving HCC growth and metastasis [36].
Furthermore, several gene variants trigger the cellular oxidative stress landscape. Defined variants such as PNPLA3 I148M, TM6SF2 E167K, and MBOAT7 rs641738 aggravate steatosis and mitochondrial dysfunction, enhancing ROS output and diminishing antioxidant capacity [33,40]. Epigenetic alterations, including microRNA dysregulation, further compromise mitochondrial biogenesis and augment oxidative injury [66].
Concurrently, antioxidant defense mechanisms are blunted in the pathogenesis of MASLD. Nuclear factor erythroid 2-related factor 2 (NRF2), representing one of the master regulators of redox homeostasis, is functionally repressed by hyper nutrition, leading to insufficient transcription of detoxifying enzymes such as superoxide dismutase 2 (SOD2), catalase (CAT), and glutathione peroxidase (GPX4) [59]. Additionally, glutathione stores are depleted by chronic oxidative challenge, and the failure in the cellular antioxidant defense systems provokes ferroptosis, an iron-dependent, peroxidation-driven cell death. This leads to a rapid decline in hepatocyte function, hepatocyte loss, and compensatory proliferation [59].
In physiological conditions, NRF2 orchestrates a tiered antioxidant defense network that counterbalances the distinct ROS generated in hepatocytes [67]. Superoxide anion, produced by the mitochondrial electron transport chain, NADPH oxidases, and other oxidases, is rapidly dismutated to hydrogen peroxide by cytosolic and mitochondrial superoxide dismutases. Additionally, catalase, glutathione peroxidases, and peroxiredoxins further reduce hydrogen peroxide and lipid hydroperoxides, using glutathione or thioredoxin as electron donors. In MASLD, chronic nutrient excess, insulin resistance, and lipotoxicity increase the generation of superoxide, hydrogen peroxide, and hydroxyl radicals beyond this buffering capacity [68]. Hyper nutrition-associated repression or maladaptive activation of NRF2 leads to inadequate induction of these detoxifying enzymes and phase II antioxidant genes. Concomitant depletion of reduced glutathione and impaired NADPH regeneration compromise the recycling of glutathione peroxidases and peroxiredoxins, thereby favoring lipid peroxidation, protein oxidation, and conditions prone to ferroptosis [69]. Antioxidant vitamins play a crucial role in these pathways. Specifically, vitamin E and other lipophilic antioxidants terminate lipid peroxyl radical chain reactions within membranes and lipid droplets, while vitamin C and other water-soluble vitamins regenerate oxidized vitamin E and support maintenance of the glutathione pool. Consequently, the antioxidant activities of vitamin C and E effectively mitigate hepatocyte injury [70].
Patients with obesity and MASLD often consume energy-dense, micronutrient-poor diets and may exhibit suboptimal circulating levels of vitamins [70]. This further weakens endogenous antioxidant defenses and promotes disease progression. Conversely, nutrient stress states such as fasting or caloric restriction transiently enhance mitochondrial ROS but concomitantly activate NRF2, AMPK, and sirtuin pathways in mice [71]. This leads to the upregulation of antioxidant and cytoprotective genes, improving redox resilience in an adaptive hormetic response that appears blunted in chronic overnutrition. Understanding how ROS species, enzymatic and non-enzymatic antioxidants, NRF2 signaling, and vitamin status interact in MASLD is crucial for the rational design of antioxidant- and nutrition-based preventive and therapeutic strategies.
Therapeutic strategies targeting oxidative stress thus constitute an attractive avenue for MASLD-HCC prevention. Lifestyle interventions, bariatric surgery, and insulin-sensitizing agents alleviate lipotoxic substrates and restore mitochondrial function [72]. Fasting-mimicking drugs, such as metformin, activate AMPK and not only reduce DNL but also enhance antioxidant defenses and increasing glucose uptake [73], whereas depletion of NRF2 or CYP2E1 protects against steatohepatitis and HCC in preclinical models [74,75]. However, these findings are still under debate, and NRF2 seems to have a dual role in HCC development [76]. Additionally, novel agents such as ferroptosis inhibitors [77], mitochondrial-targeted antioxidants such as the synthetic analogue of coenzyme Q10 known as MitoQ [78], and peroxisome proliferator-activated receptor (PPAR) agonists [79] are under investigation, aiming to interrupt the cycle of ROS-mediated injury that is a driving force of MASLD toward malignancy. Given the uncertainties surrounding our complete understanding of the molecular pathogenesis of disease modulation, the ROS-driven cycle remains pivotal in driving the increasing number of obesity-related MASLD-HCC worldwide. Innovative approaches are crucial in this regard.
Chronic, low-grade inflammation is another defining hallmark that drives the progression from lipid-laden hepatocytes in MASLD towards liver fibrosis, its hemodynamic complications, and HCC. As discussed above, steatotic hepatocytes act as both initiators and amplifiers of this inflammatory cascade by releasing DAMPs, saturated fatty acid metabolites, and triggering oxidative stress. DAMPs are recognized by pattern-recognition receptors (PRRs) such as TLR4 and nucleotide-binding oligomerization domain-like receptor family pyrin domain containing-3 (NLRP3) located on resident Kupffer cells and recruited monocyte-derived macrophages [43]. The activation of these receptors triggers downstream signaling through NF-κB and MAPKs, leading to the transcription of pro-inflammatory cytokines, reinforcing insulin resistance, stimulating the activation of hepatic stellate cells, and perpetuating hepatocellular injury.
The gut-liver axis further enhances hepatic inflammation during MASLD. High-fat, low-fiber diets promote gut dysbiosis and increased intestinal permeability, facilitating the translocation of lipopolysaccharide (LPS) into the portal circulation [80]. LPS acts as an endotoxin that binds to TLR4 on Kupffer cells and sinusoidal endothelial cells, intensifying cytokine release and ROS production [80]. Simultaneously, adipose-tissue macrophages in obese individuals secrete TNF-α and IL-6 that reach the liver via the systemic circulation, highlighting the complexity of a metabolic low-grade, chronic, subclinical systemic inflammatory state (metaflammation).
Inflammasome activation is a crucial amplifier of sterile inflammation in MASLD. Excess saturated fatty acids, cholesterol crystals, and ROS prime and activate NLRP3, leading to caspase-1-mediated cleavage of pro-IL-1β into its mature, bioactive forms [81]. These cytokines drive leukocyte recruitment to the liver and promote hepatocyte pyroptosis, releasing additional DAMPs and reinforcing the inflammatory circuit [82,83]. NLRP3-deficient mouse models exhibit attenuation of steatohepatitis and reduced HCC burden, highlighting the translational relevance of inflammasome blockade [84].
Adaptive immune responses become dysregulated as MASLD progresses. Lipid antigens presented by thymocyte antigen CD1d molecules activate hepatic invariant natural killer T (iNKT) cells, initially having protective effects through IL-4 and IL-10 secretion but later adopting a pro-fibrogenic phenotype characterized by interferon-γ (IFN-γ) and TNF-α production [85]. Th17 cell expansion and elevated IL-17 levels correlate with liver fibrosis stages and tumor growth, while cytotoxic CD8+ T cells undergo exhaustion due to persistent antigenic stimulation, up-regulation of programmed cell-death-1 (PD-1), and an immunosuppressive cytokine milieu [86]. Regulatory T cells (Tregs) accumulate in the steato-fibrotic liver, secreting transforming growth factor-β (TGF-β) and IL-10, thereby dampening anti-tumor immunity and facilitating oncogenesis [87]. This evolving immunological landscape supports the concept of “immune senescence” in MASLD-HCC, which might explain the reduced efficacy of immune-checkpoint inhibitors in this numerically increasing patient population [88].
Cytokine-driven cross talks between parenchymal and non-parenchymal cells accelerate fibrogenesis and angiogenesis. IL-6 and TNF-α activate Janus kinase (JAK)-Signal transducer and activator of transcription (STAT) pathways in hepatocytes, promoting compensatory proliferation and survival of dysplastic cell clones [89]. At the same time, TGF-β released by Kupffer cells and platelets stimulates hepatic stellate cell transdifferentiation into myofibroblasts, depositing extracellular matrix that remodels the sinusoidal architecture and promotes HIF-1α-mediated vascular endothelial growth factor (VEGF) expression. The resulting neo-vasculature supplies nutrients to emerging tumor nodules while enabling immune-cell trafficking that is skewed toward tumor-promoting subsets.
Similar to the generation of ROS, genetic factors in humans can predispose to inflammation. The PNPLA3 I148M variant hampers lipid-droplet remodeling, leading to increased lipotoxic stress and heightened cytokine production. TM6SF2 and MBOAT7 polymorphisms also exacerbate steatohepatitis through dysfunctional lipid handling and altered membrane composition [33,40]. Epigenetic modifications, including DNA methylation of anti-inflammatory genes and microRNA dysregulation such as miR-155 and miR-34a, further perpetuate a pro-inflammatory state in hepatocytes [90]. The multitude of factors impacting the inflammatory responses in MASLD illustrates the notion that this disease is triggered by a dynamic, multi-cellular network that links metabolic dysfunction, lipotoxicity, fibrogenic remodeling, and immune evasion, ultimately creating an environment conducive to hepatocarcinogenesis.
These pathogenic models pave the way for active investigation of therapeutic approaches targeting inflammatory pathways. Lifestyle modification and bariatric surgery reduce adipose-derived cytokines and improve gut barrier function. Several pharmacologic agents such as glucagon-like peptide-1 (GLP-1) receptor agonists (e.g., exenatide, dulaglutide, liraglutide, and semaglutide), PPAR agonists (e.g., clofibrate, gemfibrozil, and fenofibrate), and statins, exhibit anti-inflammatory properties alongside metabolic benefits. Specific inhibitors of NLRP3, TLR4 antagonists, and monoclonal antibodies against IL-1β or IL-17 are being explored in early-phase trials [79,91]. Notably, the dual GLP-1/GIP agonist tirzepatide and the FXR agonist obeticholic acid have demonstrated histologic improvements that correlate with reduced inflammatory markers, offering hope for disease modification [92,93].
4.4 Immunological Milieu and Immunometabolism
Obesity-associated MASLD creates a chronically inflamed, immunosuppressed hepatic microenvironment, in which innate immune cells undergo profound metabolic rewiring. Activated Kupffer cells and infiltrating monocyte-derived macrophages shift toward an M1-like, glycolysis-dominated phenotype that promotes production of TNF-α, IL-1β and IL-6, further promoting insulin resistance and hepatocyte injury [43]. However, under conditions of chronic obesity-related metabolic dysfunction, these macrophages acquire lipid droplets, upregulate PPAR-γ, and transition to an M2-like state that secretes TGF-β and VEGF, promoting hepatic fibrogenesis and neoangiogenesis [94]. Neutrophils, mobilized by chemokines such as C-X-C motif chemokine ligand-1 (CXCL1) and -2 (CXCL2), generate myeloperoxidase-derived oxidants and neutrophil extracellular traps, but prolonged exposure to lipotoxic mediators induces immunosuppressive granulocytic myeloid-derived suppressor cells that dampen anti-tumor T-cell responses [95].
Natural killer (NK) cells and invariant natural killer T (iNKT) cells play pivotal roles in tumor surveillance, mediating antitumor functions through cytotoxicity and cytokine secretion. NK cells can prevent the development of liver fibrosis by exerting cytolytic activity against hepatic stellate cells [96]. However, in experimental models of SLD, NK cells are activated and trigger a pro-inflammatory cytokine milieu, thereby promoting MASLD development [97]. Moreover, excess free fatty acids downregulate activating receptors, such as natural cytotoxicity triggering receptor (NKp46) and natural killer group 2 member D (NKG2D) and force a metabolic switch from oxidative phosphorylation to aerobic glycolysis, limiting cytolytic capacity [78]. Concurrently, increased expression of PD-1 and the T-cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT) drives an exhausted phenotype that permits clonal expansion of dysplastic hepatocytes [98].
Adaptive immunity is equally reshaped by metabolic cues. Steatotic antigen-presenting cells favor differentiation of naïve CD4+ T cells toward pro-inflammatory Th17 cells via IL-6 and IL-23. Chronic antigenic load skews CD8+ T cells toward an exhausted, PD-1+/T-cell immunoglobulin and mucin domain-containing 3 (TIM-3)+ state characterized by impaired interferon-γ production and reliance on fatty-acid oxidation for survival [99,100,101]. Tregs accumulate in advanced MASLD and early HCC, triggered by TGF-β and lipid-mediated activation of the transcription factor FOXP3, consuming local glucose, thereby establishing a metabolically restrictive and immunosuppressive niche [102].
Immunometabolism also influences non-cellular components of the tumor microenvironment. Hypoxia arising from sinusoidal capillarization activates HIF-1α, inducing glycolytic enzymes and lactate transporters in both hepatocytes and immune cells. Extracellular lactate acidifies the milieu, further suppressing effector T-cell function while stimulating increased glycolysis and lactate production, M2 polarization, and angiogenesis [94]. In parallel, cholesterol crystals and oxidized phospholipids accumulate in hepatic stellate cells and dendritic cells, triggering NLRP3 inflammasome activation and perpetuating IL-1β release [103]. Excessive intracellular cholesterol reduces membrane fluidity and blunts antigen presentation, weakening anti-tumor immunity [104]. In addition, several genetic variants discussed above aggravate steatosis and modulate immune infiltration. Specifically, the PNPLA3 I148M polymorphism promotes lipid droplet retention in hepatocytes and macrophages, increasing lipotoxic stress and cytokine production in humans. On the other hand, TM6SF2 and MBOAT7 variants change membrane lipid composition, affecting TLR signaling thresholds and inflammasome sensitivity in humans. These gene polymorphisms could impact the variability in immune responses and cancer risk among the MASLD population [33,40].
The overall impact of these changes is an immune environment characterized by a contradiction, in which increased inflammatory signals exist alongside impaired tumor immunosurveillance. This allows premalignant clones to arise, escape immune detection and thrive. This duality has significant implications, as recent studies indicate that HCC related to MASLD shows a poorer response to immune checkpoint inhibitors [105]. This may be attributed to the high presence of exhausted T cells, immunosuppressive myeloid cell populations, and anergy induced by various metabolites.
Therapeutic strategies aimed at recalibrating immunometabolism are of considerable clinical interest. Lifestyle interventions and metabolic bariatric surgery decrease circulating free fatty acids and restore mitochondrial fitness in immune cells, partially reversing exhaustion signatures. Pharmacologic activators of AMPK (e.g., metformin) and inhibitors of mTOR complex 1 (e.g., rapalogs) can re-establish oxidative metabolism, enhance memory T-cell formation, and reduce M2 macrophage polarization. Similarly, synthetic and semi-synthetic FXR agonists (e.g., obeticholic acid, fexaramine, and chenodeoxycholic acid) and GLP-1 receptor agonists indirectly modulate the immune microenvironment by reducing steatosis and gut-derived endotoxemia due to modification of the gut microbiota [106]. Moreover, novel agents that block IL-17 and TNF-α, which amplify and perpetuate liver damage resulting from NLRP3 activation, or that deplete myeloid-derived suppressor cells, in combination with immune-checkpoint inhibitors, may overcome the metabolic and immunological barriers unique to MASLD-HCC [107]. It is expected that this ever-expanding therapeutic arsenal may contribute to improving our understanding of the complex immunological milieu and immunometabolism in the field of obesity-related MASLD-HCC.
4.5 Regulation of Signaling Pathways Such as mTOR, AMPK, and NF-κB, and How They Influence Processes Like Cell Proliferation, Apoptosis, Autophagy, Migration, and Differentiation
As discussed, metabolic overload in obesity-associated MASLD disrupts nutrient-sensing and stress-response signaling networks. AMPK, mTOR, and NF-κB pathways are key regulators that integrate metabolic processes with cellular fate decisions [26,27,28]. Based on this important functionality, it is evident that these pathways can accelerate hepatocarcinogenesis when chronically dysregulated.
The multi-protein complex mTORC1, which includes the kinase mTOR, is consistently activated in steatotic hepatocytes due to excess amino acids, insulin, and growth factors [108]. This leads to the phosphorylation of S6 kinase and the eukaryotic translation initiation factor 4E-binding protein (4E-BP1), promoting cap-dependent translation of cyclin D1, c-Myc, and other drivers of G1/S progression, resulting in uncontrolled cell proliferation [108]. Persistent mTORC1 signaling also suppresses autophagy, including lipophagy, by phosphorylating UNC51-like autophagy-activating kinase 1 (ULK1) [108]. This limits the ability of the cell to clear damaged mitochondria and excess lipid droplets, leading to increased oxidative stress and genomic instability. Additionally, mTORC1 crosstalk with HIF-1α promotes glycolytic flux and VEGF expression, enhancing angiogenesis and migration of premalignant hepatocytes [109].
On the other hand, AMPK acts as an energy sensor that is typically activated by rising AMP/ATP ratios. In MASLD, lipotoxicity and insulin resistance reduce its activity, removing a critical restraint on mTORC1 and on DNL via SREBP-1c. Reduced AMPK signaling leads to diminished fatty acid oxidation and autophagic flux, allowing for triglyceride accumulation and ER stress. This weakens p53-mediated checkpoints that would otherwise induce apoptosis in damaged cells [110,111]. In line with these findings, therapeutic reactivation of AMPK through caloric restriction, exercise, or metformin restores mitochondrial biogenesis and rescues autophagy, reducing ROS, limiting mutation burden, and repressing tumor initiation in preclinical MASLD-HCC models [46]. In all these processes, NF-κB serves as a central hub linking metabolic stress to inflammatory and survival pathways [94]. DAMPs, LPS, and saturated fatty acids engage TLR4 and IL-1 receptor signaling, leading to the induction of TNF-α, IL-6, and anti-apoptotic proteins, such as B-cell lymphoma-extra-large (Bcl-xL) and cellular FLICE-inhibitory protein (c-FLIP), protecting injured hepatocytes from apoptosis, promoting survival, and sustaining compensatory proliferation and fibrosis. NF-κB also up-regulates matrix metalloproteinases (MMPs) and chemokines that facilitate extracellular matrix remodeling, cell migration, tumor invasion, and metastasis [112].
It should be noted that these pathways interact bidirectionally. mTORC1 activation inhibits autophagy, increasing ROS, which activates NF-κB, supporting survival and inflammation. Conversely, AMPK phosphorylation of TSC complex subunit 2 (TSC2 or tuberin) and regulatory associated protein of mTOR (RAPTOR) restrains mTORC1, while AMPK-mediated phosphorylation of p65 dampens NF-κB transcriptional activity, highlighting the tumor-suppressive role of AMPK [113]. Dysregulated lipid species such as diacylglycerols and ceramides modulate these crosstalk nodes by activating protein kinase C or by inhibiting AKT, tipping the balance toward either unchecked proliferation or lipotoxic apoptosis depending on the cellular context [114].
Studies in zebrafish have shown that cell differentiation is also influenced by these pathways. mTORC1 is involved in hepatocyte-to-cholangiocyte transdifferentiation and cholangiocyte-to-hepatocyte transdifferentiation [115]. It is most likely that this reprogramming during tissue injury is mediated through Notch and Wnt/β-catenin pathways, contributing to the emergence of progenitor-like HCC subtypes with aggressive behavior [116]. On the other hand, AMPK impacts histone deacetylation and gene expression, thereby contributing to the global regulation of protein acetylation and connecting epigenetic chromatin modifications with the cellular metabolic state [117]. All these processes interfere with NF-κB signaling that up-regulates epithelial-mesenchymal transition (EMT) transcription factors, such as Snail and Twist, driving loss of epithelial polarity, and further driving cell migration and invasion, thereby promoting the acquisition of invasive traits [118].
Collectively, the aberrant activation of mTOR and NF-κB coupled with AMPK deficiency orchestrates a molecular environment that promotes hepatocyte proliferation, suppresses apoptosis and autophagy, enhances migration and dedifferentiation, and drives the progression from MASLD to HCC. Consequently, the rebalancing of this signaling triad is crucial for reducing the increasing burden of obesity-related HCC and pharmacological modulation of these axes is a rational strategy for MASLD-HCC prevention and therapy. mTOR inhibitors like everolimus show anti-proliferative effects but are limited by feedback activation of AKT. Dual PI3K/mTOR or mTORC1/2 inhibitors, and intermittent dosing schedules, are being explored to mitigate resistance [119]. AMPK activators, including metformin and the mitochondrial complex I inhibitor Imeglimin, are consolidated approaches or emerging therapies for the management of diabetes [120,121]. It is logical to speculate that the combination of these drugs with immune checkpoint inhibitors (ICIs) will be potentially effective in counteracting hepatic carcinogenesis, especially of MASLD-related HCC. Moreover, NF-κB blockade through IKKβ inhibitors or proteasome inhibitors can restore apoptotic sensitivity. However, systemic immunosuppression remains a concern [122]. Precision approaches that target liver-specific NF-κB components or upstream sensors, such as TLR4 antagonists and NLRP3 inhibitors, may offer safer alternatives.
As discussed earlier, the metabolic dysfunction associated with obesity-associated MASLD requires that hepatocytes and non-parenchymal cells persistently experience lipotoxicity and oxidative stress. Physiologically, the metabolic zonation of the liver is an important feature that helps maintain whole body homeostasis [123]. However, during progressive MASLD zonation is disturbed, leading to zones of micro-hypoxia that act as powerful inducers of intra- and extrahepatic neoangiogenesis [124]. Accumulated fat droplets enlarge hepatocytes, compress sinusoidal spaces, and disrupt liver perfusion, stabilizing transcription factors HIF-1α and HIF-2α, which up-regulate vascular endothelial growth factor-A (VEGF-A), angiopoietin-2, and platelet-derived growth factor-BB (PDGF-BB). These mediators promote the sprouting of aberrant neovessels that, while initially compensating for oxygen deprivation, will ultimately provide a vascular scaffold supporting the clonal expansion of dysplastic hepatocytes [125]. Kupffer cells and infiltrating monocyte-derived macrophages amplify this response by producing TNF-α and IL-6, cytokines that further enhance HIF-1α activity and stimulate VEGF secretion. Meanwhile, ROS generated by dysfunctional mitochondria oxidize lipids and activate NF-κB, further driving proangiogenic gene expression. Activated hepatic stellate cells triggered by TGF-β and lipotoxic aldehydes, transdifferentiate to myofibroblasts and secrete type I collagen and laminin, promote the capillarization of sinusoids, narrowing the fenestrations of liver sinusoidal endothelial cells (LSECs), and promoting hypoxia, establishing a self-perpetuating loop of liver fibrosis and neoangiogenesis [65,126]. Under these conditions, lipid-laden LSECs lose their anti-angiogenic phenotype and express adhesion molecules like vascular cell adhesion molecule 1 (VCAM-1) and intracellular adhesion molecule 1 (ICAM-1), facilitating recruitment of proangiogenic neutrophils and myeloid-derived suppressor cells (Fig. 2) [127].

Figure 2: Redox-inflammation-immunometabolism feedback loops that enable fibrogenesis, angiogenesis, and immune escape in obesity-related MASLD-HCC. (A) Caloric excess and lipotoxicity increase reactive oxygen species (ROS) production from mitochondria (electron leak at complexes I/III), peroxisomal β-oxidation of very-long-chain fatty acids, and failure in endoplasmic reticulum (ER) protein folding and unfolded protein response (UPR). The resulting superoxide and hydrogen peroxide initiate lipid peroxidation and DNA damage. (B) Damage-associated molecular patterns (DAMPs), lipopolysaccharide (LPS)/TLR4 signaling, and cholesterol crystals/ROS prime and activate NLRP3, leading to caspase-1 activation, IL-1β/IL-18 release, and pyroptosis, which amplify hepatocyte injury and stellate cell activation. (C) Kupffer cells/monocyte-derived macrophages shift from glycolytic M1-like activation toward lipid-laden M2-like states that secrete TGF-β/VEGF. NK and CD8+ T cells downregulate cytolytic programs and exhibit PD-1/TIGIT-driven exhaustion. Tregs and Th17 cells accumulate, tipping the balance toward tumor promotion. (D) Reduced AMPK activity and sustained mTORC1/NF-κB signaling suppress autophagy, promote survival/proliferation, and upregulate HIF-1α-dependent glycolysis and cytokines, locking tissues into a pro-tumor metabolic and inflammatory state. (E) Sinusoidal capillarization and hypoxia stabilize HIF-1α/2α in hepatocytes and non-parenchymal cells, inducing VEGF-A and angiopoietins. The resulting tortuous, leaky neovessels facilitate tumor growth, immune evasion, and heterogeneous drug delivery. (F) Lifestyle interventions, bariatric/metabolic surgery, AMPK activators (e.g., metformin), rapalogs, NLRP3/TLR4 antagonists, GLP-1/FXR agonists, ferroptosis modulators, and anti-angiogenics (e.g., anti-VEGF) target different nodes in the loop. Additionally, a rational combination of these strategies may be helpful in some cases in MASLD-HCC therapy. This original illustration was created using Servier Medical ART (SMART) and is licensed under the Creative Commons Attribution 4.0 International License (CC BY 4.0).
Moreover, the newly formed vessels in tumors are structurally and functionally abnormal, being tortuous, poorly covered by pericytes, and hyper-permeable, leading to irregular perfusion and a heterogeneous tumor microenvironment [128]. This “vascular chaos” promotes the selection of aggressive clones, facilitates metastatic dissemination, and hinders drug delivery. The metabolic environment rich in free fatty acids and lactate impairs dendritic cell maturation and promotes immune tolerance. This partially explains why immunotherapy appears to be less effective in MASLD-HCC compared to HCC arising in the context of chronic liver disease due to viral etiology [105].
Nevertheless, angiogenesis in obesity-driven MASLD represents the vascular arm of a broader maladaptive response to lipotoxic hypoxia. It integrates signals from nutrient-sensing pathways, innate immunity, and fibrogenesis to create a pro-tumor niche. Because neovascularization promotes the expansion of malignant hepatocytes and shapes an immune-evasive microenvironment, successful interception of the obesity-MASLD-HCC cascade will require strategies that simultaneously target metabolic dysfunction, suppress inflammation, reduce portal pressure, and remodel aberrant vasculature [129].
Therapeutically, angiogenesis is a prime target in advanced diseases. Bevacizumab, an anti-VEGF monoclonal antibody, combined with the programmed death-ligand-1 inhibitor atezolizumab, is a first-line regimen for the treatment of patients with unresectable HCC [130]. However, the efficacy of this immunotherapy in HCC with non-viral etiologies such as MASLD is less effective, possibly because VEGF blockade does not fully reverse the immune-metabolic suppression characteristic of this etiology [131]. Multi-kinase inhibitors such as sorafenib, lenvatinib, and regorafenib simultaneously inhibit VEGF receptors, PDGF receptors, and RAF kinases. However, their benefits are counterbalanced by significant adverse effects, especially in obese patients with metabolic comorbidities [132]. Combination approaches that associate anti-angiogenic agents with AMPK activators or NLRP3 inflammasome inhibitors might be a rational strategy for simultaneously correcting both vascular and inflammatory derangements, although this needs to be explored experimentally.
Treatment options for obesity and MASLD have been extensively discussed elsewhere [11,133]. Here, we will specifically focus on the challenges posed by obesity-related MASLD-HCC. These challenges include delayed diagnosis resulting from insufficient surveillance, limited access to curative interventions such as tumor ablation, surgical resection, and liver transplantation, as well as diminished responsiveness to certain treatments used in advanced disease states. These treatments include locoregional therapies like transarterial chemoembolization (TACE) and transarterial radioembolization (TARE), as well as systemic therapies like ICIs such as atezolizumab and durvalumab, often used in combination with other drugs like bevacizumab or tremelimumab, and tyrosine kinase inhibitors (TKIs) such as sorafenib and regorafenib.
MASLD-HCC is often detected at later stages because surveillance programs do not consistently include patients without cirrhosis [134]. Many patients are not included in surveillance programs, leading to diagnosis at more advanced stages (Barcelona Clinic Liver Cancer C or D), which limits options for curative treatment [135].
5.2 Limited Access to Curative Treatment
Patients with MASLD-HCC have a higher risk of post-surgical complications, including increased morbidity and mortality, which may limit eligibility for surgical resection [136]. The presence of the full-blown metabolic syndrome negatively impacts post-operative outcomes [137], and malnutrition, commonly observed in this patient population, is a further contributor to increased post-operative risk.
Nutrition plays a critical yet often overlooked role in the management of MASLD-cirrhosis. Addressing nutritional deficiencies is particularly important for patients with MASLD-HCC who are awaiting interventions to slow disease progression or are being prepared for curative treatments such as surgical resection or liver transplantation [138].
5.4 Metabolic Bariatric Surgery
There are no clear guidelines on the use of metabolic bariatric surgery in patients with MASLD-cirrhosis awaiting liver transplantation. Furthermore, patients with MASLD-HCC have a higher mortality rate on the waiting list [139]. The possible utilization of metabolic bariatric surgery in achieving hepatic recompensation among those with MASLD-cirrhosis is gaining increasing attention [140]. While sleeve gastrectomy can improve candidacy in morbidly obese patients awaiting transplantation [141], it remains to be assessed whether MBS is a reasonable option among well-selected individuals with compensated MASLD-HCC and without significant portal hypertension.
Accumulating evidence indicates that chronic sterile low-grade inflammation of metabolic origin in the context of MASLD serves not only as the primary trigger of malignant transformation of hepatocyte lineage, but also reshapes the immune background. This leads to immune system exhaustion and favors the loss of cancer immune surveillance [142]. These biological phenomena explain why immunotherapy may be less effective in patients with MASLD-related HCC compared to those with HCC due to other etiologies, owing to immune alterations specific to MASLD [105].
In summary, obesity-related MASLD-HCC presents unique clinical challenges. These challenges include frequent late-stage diagnoses and limited availability of curative interventions such as surgical resection or transplantation. Advanced therapeutic modalities, like TACE, transarterial radioembolization (TARE), immune checkpoint inhibitors, and tyrosine kinase inhibitors, often show reduced efficacy in this population. The lack of adequate surveillance, especially in non-cirrhotic individuals, leads to delayed detection and limits treatment options. Metabolic syndrome and malnutrition increase perioperative risk and hinder postoperative recovery. However, nutritional optimization before major interventions is often overlooked and remains essential. Dietary approaches, such as intermittent fasting, may improve insulin sensitivity and reduce hepatic steatosis, potentially impacting tumor metabolism and immune response, although long-term safety data are scarce. Current guidelines on bariatric surgery in MASLD-cirrhosis transplant candidates are unclear, but sleeve gastrectomy may offer advantages in specific cases. The use of metabolic surgery in compensated MASLD-HCC is still being studied. Furthermore, chronic inflammation from metabolic dysfunction impairs the effectiveness of immunotherapy by promoting immune exhaustion and reducing cancer surveillance.
6 Conclusions and Future Direction
Patients with obesity-related MASLD-HCC often present with advanced-stage disease, multiple comorbidities, and increased risk of post-operative complications. To counteract this unfavorable clinical scenario, the research agenda for MASLD-HCC prioritizes improved risk stratification [143] and early disease detection through the development of enhanced screening and surveillance methods.
In line with emerging expert suggestions, surveillance may be extended—beyond cirrhosis—to a portion of the elderly with MASLD who have non-invasively identified advanced fibrosis [6,144,145,146]. In this context, integrated risk scores that combine metabolic risk factors, genetic information, and non-invasive fibrosis markers could be implemented to identify high-risk patients who should undergo HCC surveillance with an improved cost-benefit ratio. Blood-based biomarkers, imaging advancements, and artificial intelligence (AI)-assisted risk prediction tools offer the potential to enhance HCC surveillance in MASLD.
Key areas of investigation include understanding the specific molecular mechanisms and tumor microenvironment characteristics of (obesity-related)-MASLD-HCC, refining non-invasive diagnostic techniques, and identifying new therapeutic targets. Advancing innovative therapies like nanotechnologies and personalized medicine, which were not discussed in this review, is crucial, especially considering the limited effectiveness of currently available immune checkpoint inhibitors. Additionally, there are fewer approved pharmacological treatments for MASLD-HCC compared to HCC from other causes, highlighting the significance of supportive management strategies like weight reduction, increased physical activity, improved nutrition, comprehensive management of dysmetabolic conditions (such as type 2 diabetes, dyslipidemia, and arterial hypertension), and portal hypertension in caring for patients with obesity-related MASLD-HCC.
Ongoing clinical trials are anticipated to address the unique pathophysiology of MASLD-related HCC to optimize treatment strategies. Simultaneously, research efforts should concentrate on developing new agents that target MASH/MASLD directly, such as resmetirom and semaglutide [147,148], even though their roles in MASLD-HCC are still unclear.
Acknowledgement:
Funding Statement: The authors received no specific funding for this study.
Author Contributions: Manuscript preparation, review and editing, visualization, supervision: Ralf Weiskirchen, Amedeo Lonardo; draft manuscript preparation: Ralf Weiskirchen, Amedeo Lonardo; review and editing: Ralf Weiskirchen, Amedeo Lonardo; visualization: Ralf Weiskirchen, Amedeo Lonardo; supervision: Ralf Weiskirchen, Amedeo Lonardo. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report.
List of Abbreviations Used
| 4E-BP1 | Eukaryotic translation initiation factor 4E-binding protein |
| 4-HNE | 4-hydroxynonenal |
| ACC | Acetyl-CoA carboxylase |
| AKT | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| BCLC | Barcelona Classification of Liver Cancer |
| BMI | Body mass index |
| CAT | Catalase |
| ChREBP | Carbohydrate-responsive element-binding protein |
| CLD | Chronic liver disease |
| CYP2E1 | Cytochrome P450 2E1 |
| CXCL1/2 | C-X-C motif chemokine ligand-1/2 |
| DALY | Disability-adjusted life year |
| DAMP(s) | Danger-associated molecular pattern(s) |
| DNL | De novo lipogenesis |
| EMT | Epithelial–mesenchymal transition |
| ER | Endoplasmic reticulum |
| FOXP3 | Forkhead-box-protein 3 |
| FPG | Fasting plasma glucose |
| FXR | Farnesoid X receptor |
| GCKR | Glucokinase regulator protein |
| GPX4 | Glutathione peroxidase |
| HCC | Hepatocellular carcinoma |
| HIF-1α | Hypoxia-inducible factor-1α |
| HMG-CoA | 3-hydroxy-3-methyl-glutaryl-CoA |
| HSC(s) | Hepatic stellate cell(s) |
| ICAM | Intercellular adhesion molecule 1 |
| ICI(s) | Immune checkpoint inhibitor(s) |
| iNKT | Invariant natural killer T |
| IL | Interleukin |
| IFN-γ | Interferon-γ |
| JNK | c-Jun N-terminal kinase |
| LPS | Lipopolysaccharides |
| LSEC(s) | Liver sinusoidal endothelial cell(s) |
| MAFLD | Metabolic dysfunction-associated fatty liver disease |
| MAPK(s) | Mitogen-activated protein kinase(s) |
| MASH | Metabolic dysfunction-associated steatohepatitis |
| MASLD | Metabolic dysfunction-associated steatotic liver disease |
| MASLD-HCC | Metabolic dysfunction-associated steatotic liver disease-related hepatocellular carcinoma |
| MBOAT7 | Membrane-bound O-acetyltransferase domain-containing protein 7 |
| MBS | Metabolic bariatric surgery |
| MDA | Malondialdehyde |
| MDSC(s) | Myeloid-derived suppressor cell(s) |
| mTOR | Mechanistic target of rapamycin |
| mTORC1 | Mechanistic target of rapamycin complex 1 |
| NAFLD | Nonalcoholic fatty liver disease |
| NHANES | National Health and Nutrition Examination Survey |
| NF-κB | Nuclear factor-κB |
| NLRP3 | Nucleotide-binding oligomerization domain-like receptor family pyrin domain containing-3 |
| NOX | NADPH oxidase |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| PD-1 | Programmed cell-death-1 |
| PD-L1 | Programmed death-ligand-1 |
| PDGF-BB | Platelet-derived growth factor-BB |
| PLC | Primary liver cancer |
| PNPLA3 | Patatin-like phospholipase domain-containing 3 |
| PPAR | Peroxisome proliferator-activated receptor |
| PRR(s) | Pattern-recognition receptor(s) |
| RAPTOR | Regulatory-associated protein of mTOR |
| ROS | Reactive oxygen species |
| S6K | Ribosomal protein-S6 kinase |
| SDI | Socio-demographic index |
| SLD | Steatotic liver disease |
| SOD2 | Superoxide dismutase 2 |
| SREBP-1c | Sterol regulatory element-binding protein-1c |
| TACE | Transarterial chemoembolization |
| TARE | Transarterial radioembolization |
| TGF-β | Transforming growth factor-β |
| TIGIT | T-cell immunoreceptor with Ig and ITIM domains |
| TIM3 | T-cell immunoglobulin and mucin-domain containing-3 |
| TKI(s) | Tyrosine kinase inhibitors |
| TLR(s) | Toll-like receptor(s) |
| TM6SF2 | Transmembrane 6 superfamily, member 2 |
| TNF-α | Tumor necrosis factor-α |
| Tregs | Regulatory T cells |
| ULK | UNC-51-like autophagy-activating kinase 1 |
| UPR | Unfolded protein response |
| VCAM | Vascular cell adhesion molecule 1 |
| VEGF | Vascular endothelial growth factor |
| XO | Xanthine oxidase |
References
1. World Health Organization . Obesity and overweight [Internet]. [cited 2025 Nov 14]. Available from: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. [Google Scholar]
2. Lonardo A , Weiskirchen R . Liver and obesity: a narrative review. Explor Med. 2025; 6: 1001334. doi:10.37349/emed.2025.1001334. [Google Scholar] [CrossRef]
3. Callahan EA . The science, strengths, and limitations of body mass index. In: National Academies of Sciences, Engineering and Medicine, Health and Medicine Division, Food and Nutrition Board, Roundtable on Obesity Solutions, Callahan EA , editors. Translating knowledge of foundational drivers of obesity into practice: proceedings of a workshop series. Washington, DC, USA: National Academies Press; 2023. p. 99– 106. [Google Scholar]
4. Ross R , Neeland IJ , Yamashita S , Shai I , Seidell J , Magni P , et al. Waist circumference as a vital sign in clinical practice: a Consensus Statement from the IAS and ICCR Working Group on Visceral Obesity. Nat Rev Endocrinol. 2020; 16( 3): 177– 89. doi:10.1038/s41574-019-0310-7. [Google Scholar] [CrossRef]
5. Rubino F , Cummings DE , Eckel RH , Cohen RV , Wilding JPH , Brown WA , et al. Definition and diagnostic criteria of clinical obesity. Lancet Diabetes Endocrinol. 2025; 13( 3): 221– 62. Erratum in: Lancet Diabetes Endocrinol. 2025;13(3):e6. doi:10.1016/S2213-8587(25)00006-3. [Google Scholar] [CrossRef]
6. Tacke F , Horn P , Wai-Sun Wong V , Ratziu V , Bugianesi E , Francque S , et al. EASL–EASD–EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J Hepatol. 2024; 81( 3): 492– 542. doi:10.1016/j.jhep.2024.04.031. [Google Scholar] [CrossRef]
7. Lonardo A , Zheng MH , Eslam M . MASLD vs. MAFLD. A narrative review. Explor Dig Dis. 2025; 4: 100586. doi:10.37349/edd.2025.100586. [Google Scholar] [CrossRef]
8. Asafo-Agyei KO , Samant H . Hepatocellular carcinoma. Treasure Island, FL, USA: StatPearls Publishing; 2025. [Google Scholar]
9. Lugari S , Baldelli E , Lonardo A . Metabolic primary liver cancer in adults: risk factors and pathogenic mechanisms. Metab Target Organ Damage. 2023; 3( 1): 5. doi:10.20517/mtod.2022.38. [Google Scholar] [CrossRef]
10. Benhammou JN , Lin J , Aby ES , Markovic D , Raman SS , Lu DS , et al. Nonalcoholic fatty liver disease-related hepatocellular carcinoma growth rates and their clinical outcomes. Hepatoma Res. 2021; 7: 70. doi:10.20517/2394-5079.2021.74. [Google Scholar] [CrossRef]
11. Ahmed SK , Mohammed RA . Obesity: prevalence, causes, consequences, management, preventive strategies and future research directions. Metab Open. 2025; 27: 100375. doi:10.1016/j.metop.2025.100375. [Google Scholar] [CrossRef]
12. Zhou XD , Chen QF , Yang W , Zuluaga M , Targher G , Byrne CD , et al. Burden of disease attributable to high body mass index: an analysis of data from the Global Burden of Disease Study 2021. eClinicalMedicine. 2024; 76: 102848. Erratum in: eClinicalMedicine. 2024;78:102958. doi:10.1016/j.eclinm.2024.102958. [Google Scholar] [CrossRef]
13. Wu C , Targher G , Byrne CD , Mao Y , Cheung TT , Yilmaz Y , et al. Global, regional, and national burden of primary liver cancer attributable to metabolic risks: an analysis of the global burden of disease study 1990-2021. Am J Gastroenterol. 2025; 120( 10): 2280– 90. doi:10.14309/ajg.0000000000003288. [Google Scholar] [CrossRef]
14. World Obesity . Economic impact of overweight and obesity set to reach 3.3% of global GDP by 2060 [Internet]. [cited 2025 Nov 14]. Available from: https://www.worldobesity.org/news/economic-cost-of-overweight-and-obesity-set-to-reach-3.3-of-global-gdp-by-2060#:~:text=GDP%20by%202060-,Economic%20impact%20of%20overweight%20and%20obesity%20set%20to%20reach,of%20global%20GDP%20by%202060&text=News%20Facts:,India%20(nearly%20$850%20billion). [Google Scholar]
15. Lonardo A . The heterogeneity of metabolic syndrome presentation and challenges this causes in its pharmacological management: a narrative review focusing on principal risk modifiers. Expert Rev Clin Pharmacol. 2023; 16( 10): 891– 911. doi:10.1080/17512433.2023.2259306. [Google Scholar] [CrossRef]
16. Lonardo A , Zheng MH , Weiskirchen R . Food insecurity is an emerging risk factor for liver disease: a scoping review. Expert Rev Gastroenterol Hepatol. 2025; 19( 9): 1033– 49. doi:10.1080/17474124.2025.2545812. [Google Scholar] [CrossRef]
17. Wong RJ . Epidemiology of metabolic dysfunction-associated steatotic liver disease (MASLD) and alcohol-related liver disease (ALD). Metab Target Organ Damage. 2024; 4: 35. doi:10.20517/mtod.2024.57. [Google Scholar] [CrossRef]
18. Paik JM , Hobbs K , Gupta A , Alkalbani RJ , Reyes MA , Younossi ZM . Prevalence of MASLD, met-ALD, and ALD and associated fibrosis among US adults: insights from NHANES 2017 to 2023. J Clin Gastroenterol. 2025. doi:10.1097/MCG.0000000000002202. [Google Scholar] [CrossRef]
19. Owrangi S , Paik JM , Golabi P , de Avila L , Hashida R , Nader A , et al. Meta-analysis: global prevalence and mortality of cirrhosis in metabolic dysfunction-associated steatotic liver disease. Aliment Pharmacol Ther. 2025; 61( 3): 433– 43. doi:10.1111/apt.18451. [Google Scholar] [CrossRef]
20. Kan C , Zhang K , Wang Y , Zhang X , Liu C , Ma Y , et al. Global burden and future trends of metabolic dysfunction-associated Steatotic liver disease: 1990–2021 to 2045. Ann Hepatol. 2025; 30( 2): 101898. doi:10.1016/j.aohep.2025.101898. [Google Scholar] [CrossRef]
21. Huang DQ , Terrault NA , Tacke F , Gluud LL , Arrese M , Bugianesi E , et al. Global epidemiology of cirrhosis—aetiology, trends and predictions. Nat Rev Gastroenterol Hepatol. 2023; 20( 6): 388– 98. doi:10.1038/s41575-023-00759-2. [Google Scholar] [CrossRef]
22. Younossi ZM , Paik JM , Lazarus JV , Burra P , Eguchi Y , Tacke F , et al. Projected global clinical, humanistic, and economic impact of metabolic dysfunction-associated steatohepatitis (MASH): the cost of inaction based on data from nine countries. Clin Gastroenterol Hepatol. 2025. doi:10.1016/j.cgh.2025.09.002. [Google Scholar] [CrossRef]
23. Golabi P , Otgonsuren M , de Avila L , Sayiner M , Rafiq N , Younossi ZM . Components of metabolic syndrome increase the risk of mortality in nonalcoholic fatty liver disease (NAFLD). Medicine. 2018; 97( 13): e0214. doi:10.1097/MD.0000000000010214. [Google Scholar] [CrossRef]
24. Wang B , Xiong Y , Huang N , Li J , Zhang S . Global, regional, and national burden of primary liver cancer among middle-aged and elderly populations from 2000 to 2021 with a prediction to 2050: a cross-sectional analysis from Global Burden of Disease Study 2021. Int J Surg. 2025; 111( 9): 5914– 26. doi:10.1097/JS9.0000000000002752. [Google Scholar] [CrossRef]
25. Mir MM , Jeelani M , Alharthi MH , Rizvi SF , Sohail SK , Wani JI , et al. Unraveling the mystery of insulin resistance: from principle mechanistic insights and consequences to therapeutic interventions. Int J Mol Sci. 2025; 26( 6): 2770. doi:10.3390/ijms26062770. [Google Scholar] [CrossRef]
26. Jiang F , Li M , Yao T , Yi X , Gao H . Research progress on AMPK in the pathogenesis and treatment of MASLD. Front Immunol. 2025; 16: 1558041. doi:10.3389/fimmu.2025.1558041. [Google Scholar] [CrossRef]
27. Cheng Z , Chu H , Seki E , Lin R , Yang L . Hepatocyte programmed cell death: the trigger for inflammation and fibrosis in metabolic dysfunction-associated steatohepatitis. Front Cell Dev Biol. 2024; 12: 1431921. doi:10.3389/fcell.2024.1431921. [Google Scholar] [CrossRef]
28. Chen G , Zhang Y , Zhou Y , Luo H , Guan H , An B . Targeting the mTOR pathway in hepatocellular carcinoma: the therapeutic potential of natural products. J Inflamm Res. 2024; 17: 10421– 40. doi:10.2147/JIR.S501270. [Google Scholar] [CrossRef]
29. Jones JG . Some paradoxes and unresolved aspects of hepatic de novo lipogenesis. npj Metab Health Dis. 2024; 2: 18. doi:10.1038/s44324-024-00020-7. [Google Scholar] [CrossRef]
30. Rao G , Peng X , Li X , An K , He H , Fu X , et al. Unmasking the enigma of lipid metabolism in metabolic dysfunction-associated steatotic liver disease: from mechanism to the clinic. Front Med. 2023; 10: 1294267. doi:10.3389/fmed.2023.1294267. [Google Scholar] [CrossRef]
31. Mathiowetz AJ , Olzmann JA . Lipid droplets and cellular lipid flux. Nat Cell Biol. 2024; 26( 3): 331– 45. doi:10.1038/s41556-024-01364-4. [Google Scholar] [CrossRef]
32. Saliba-Gustafsson P , Härdfeldt J , Pedrelli M , Parini P . Genomic signatures of MASLD: how genomics is redefining our understanding of metabolic liver disease. Int J Mol Sci. 2025; 26( 22): 10881. doi:10.3390/ijms262210881. [Google Scholar] [CrossRef]
33. Weiskirchen R , Lonardo A . PNPLA3 as a driver of steatotic liver disease: navigating from pathobiology to the clinics via epidemiology. J Transl Genet Genom. 2024; 8( 4): 355– 77. doi:10.20517/jtgg.2024.70. [Google Scholar] [CrossRef]
34. Prill S , Caddeo A , Baselli G , Jamialahmadi O , Dongiovanni P , Rametta R , et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci Rep. 2019; 9( 1): 11585. doi:10.1038/s41598-019-47737-w. [Google Scholar] [CrossRef]
35. Lonardo A , Weiskirchen R . Insulin resistance at the crossroads of metabolic inflammation, cardiovascular disease, organ failure and cancer. Biomolecules. 2025; 15( 12): 1745. doi:10.3390/biom15121745. [Google Scholar] [CrossRef]
36. Chu Q , Gu X , Zheng Q , Zhu H . Regulatory mechanism of HIF-1α and its role in liver diseases: a narrative review. Ann Transl Med. 2022; 10( 2): 109. doi:10.21037/atm-21-4222. [Google Scholar] [CrossRef]
37. Luukkonen PK , Zhou Y , Nidhina Haridas PA , Dwivedi OP , Hyötyläinen T , Ali A , et al. Impaired hepatic lipid synthesis from polyunsaturated fatty acids in TM6SF2 E167K variant carriers with NAFLD. J Hepatol. 2017; 67( 1): 128– 36. doi:10.1016/j.jhep.2017.02.014. [Google Scholar] [CrossRef]
38. Lonardo A , Targher G . Not all fat is alike in MASLD. J Hepatol. 2025; 82( 5): e271– 2. doi:10.1016/j.jhep.2024.12.025. [Google Scholar] [CrossRef]
39. Luukkonen PK , Zhou Y , Hyötyläinen T , Leivonen M , Arola J , Orho-Melander M , et al. The MBOAT7 variant rs641738 alters hepatic phosphatidylinositols and increases severity of non-alcoholic fatty liver disease in humans. J Hepatol. 2016; 65( 6): 1263– 5. doi:10.1016/j.jhep.2016.07.045. [Google Scholar] [CrossRef]
40. Chandrasekaran P , Weiskirchen R . The pivotal role of the membrane-bound O-acyltransferase domain containing 7 in non-alcoholic fatty liver disease. Livers. 2024; 4( 1): 1– 14. doi:10.3390/livers4010001. [Google Scholar] [CrossRef]
41. Teske BF , Wek SA , Bunpo P , Cundiff JK , McClintick JN , Anthony TG , et al. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell. 2011; 22( 22): 4390– 405. doi:10.1091/mbc.E11-06-0510. [Google Scholar] [CrossRef]
42. Leamy AK , Egnatchik RA , Young JD . Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog Lipid Res. 2013; 52( 1): 165– 74. doi:10.1016/j.plipres.2012.10.004. [Google Scholar] [CrossRef]
43. Xu L , Liu W , Bai F , Xu Y , Liang X , Ma C , et al. Hepatic macrophage as a key player in fatty liver disease. Front Immunol. 2021; 12: 708978. doi:10.3389/fimmu.2021.708978. [Google Scholar] [CrossRef]
44. Panwar V , Singh A , Bhatt M , Tonk RK , Azizov S , Raza AS , et al. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct Target Ther. 2023; 8( 1): 375. doi:10.1038/s41392-023-01608-z. [Google Scholar] [CrossRef]
45. Fang C , Pan J , Qu N , Lei Y , Han J , Zhang J , et al. The AMPK pathway in fatty liver disease. Front Physiol. 2022; 13: 970292. doi:10.3389/fphys.2022.970292. [Google Scholar] [CrossRef]
46. Matter MS , Decaens T , Andersen JB , Thorgeirsson SS . Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol. 2014; 60( 4): 855– 65. doi:10.1016/j.jhep.2013.11.031. [Google Scholar] [CrossRef]
47. Gunn PJ , Pramfalk C , Millar V , Cornfield T , Hutchinson M , Johnson EM , et al. Modifying nutritional substrates induces macrovesicular lipid droplet accumulation and metabolic alterations in a cellular model of hepatic steatosis. Physiol Rep. 2020; 8( 13): e14482. doi:10.14814/phy2.14482. [Google Scholar] [CrossRef]
48. Dempsey JL , Ioannou GN , Carr RM . Mechanisms of lipid droplet accumulation in steatotic liver diseases. Semin Liver Dis. 2023; 43( 4): 367– 82. doi:10.1055/a-2186-3557. [Google Scholar] [CrossRef]
49. Kristiansen MNB , Veidal SS , Christoffersen C , Jelsing J , Rigbolt KTG . Molecular characterization of microvesicular and macrovesicular steatosis shows widespread differences in metabolic pathways. Lipids. 2019; 54( 1): 109– 15. doi:10.1002/lipd.12121. [Google Scholar] [CrossRef]
50. Weiskirchen R , Lonardo A . Sex hormones and metabolic dysfunction-associated steatotic liver disease. Int J Mol Sci. 2025; 26( 19): 9594. doi:10.3390/ijms26199594. [Google Scholar] [CrossRef]
51. Mercola J . Reductive stress and mitochondrial dysfunction: the hidden link in chronic disease. Free Radic Biol Med. 2025; 233: 118– 31. doi:10.1016/j.freeradbiomed.2025.03.029. [Google Scholar] [CrossRef]
52. Chen Z , Tian R , She Z , Cai J , Li H . Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med. 2020; 152: 116– 41. Erratum in: Free Radic Biol Med. 2021;162:174. doi:10.1016/j.freeradbiomed.2020.06.011. [Google Scholar] [CrossRef]
53. Bhattarai KR , Riaz TA , Kim HR , Chae HJ . The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp Mol Med. 2021; 53( 2): 151– 67. doi:10.1038/s12276-021-00560-8. [Google Scholar] [CrossRef]
54. Lu Q , Zong W , Zhang M , Chen Z , Yang Z . The overlooked transformation mechanisms of VLCFAs: peroxisomal β-oxidation. Agriculture. 2022; 12( 7): 947. doi:10.3390/agriculture12070947. [Google Scholar] [CrossRef]
55. He F , Ge X , Liu X . Endoplasmic reticulum oxidoreductin 1α as a potential therapeutic target in diseases: from oxidative protein folding to pathophysiological mechanisms. Front Pharmacol. 2025; 16: 1709284. doi:10.3389/fphar.2025.1709284. [Google Scholar] [CrossRef]
56. Sharma S , Le Guillou D , Chen JY . Cellular stress in the pathogenesis of nonalcoholic steatohepatitis and liver fibrosis. Nat Rev Gastroenterol Hepatol. 2023; 20( 10): 662– 78. doi:10.1038/s41575-023-00832-w. [Google Scholar] [CrossRef]
57. Flessa CM , Kyrou I , Nasiri-Ansari N , Kaltsas G , Papavassiliou AG , Kassi E , et al. Endoplasmic reticulum stress and autophagy in the pathogenesis of non-alcoholic fatty liver disease (NAFLD): current evidence and perspectives. Curr Obes Rep. 2021; 10( 2): 134– 61. doi:10.1007/s13679-021-00431-3. [Google Scholar] [CrossRef]
58. Martín-Fernández M , Arroyo V , Carnicero C , Sigüenza R , Busta R , Mora N , et al. Role of oxidative stress and lipid peroxidation in the pathophysiology of NAFLD. Antioxidants. 2022; 11( 11): 2217. doi:10.3390/antiox11112217. [Google Scholar] [CrossRef]
59. Capelletti MM , Manceau H , Puy H , Peoc’h K . Ferroptosis in liver diseases: an overview. Int J Mol Sci. 2020; 21( 14): 4908. doi:10.3390/ijms21144908. [Google Scholar] [CrossRef]
60. Li J , Bai L , Xin Z , Song J , Chen H , Song X , et al. TERT-TP53 mutations: a novel biomarker pair for hepatocellular carcinoma recurrence and prognosis. Sci Rep. 2025; 15( 1): 3620. Erratum in: Sci Rep. 2025;15(1):20340. doi:10.1038/s41598-025-87545-z. [Google Scholar] [CrossRef]
61. Abuetabh Y , Wu HH , Chai C , Al Yousef H , Persad S , Sergi CM , et al. DNA damage response revisited: the p53 family and its regulators provide endless cancer therapy opportunities. Exp Mol Med. 2022; 54( 10): 1658– 69. doi:10.1038/s12276-022-00863-4. [Google Scholar] [CrossRef]
62. Shreya S , Grosset CF , Jain BP . Unfolded protein response signaling in liver disorders: a 2023 updated review. Int J Mol Sci. 2023; 24( 18): 14066. doi:10.3390/ijms241814066. [Google Scholar] [CrossRef]
63. Chen Y , Shen YQ . Role of reactive oxygen species in regulating epigenetic modifications. Cell Signal. 2025; 125: 111502. doi:10.1016/j.cellsig.2024.111502. [Google Scholar] [CrossRef]
64. Slevin E , Baiocchi L , Wu N , Ekser B , Sato K , Lin E , et al. Kupffer cells: inflammation pathways and cell-cell interactions in alcohol-associated liver disease. Am J Pathol. 2020; 190( 11): 2185– 93. doi:10.1016/j.ajpath.2020.08.014. [Google Scholar] [CrossRef]
65. Acharya P , Chouhan K , Weiskirchen S , Weiskirchen R . Cellular mechanisms of liver fibrosis. Front Pharmacol. 2021; 12: 671640. doi:10.3389/fphar.2021.671640. [Google Scholar] [CrossRef]
66. Erceg S , Munjas J , Sopić M , Tomašević R , Mitrović M , Kotur-Stevuljević J , et al. Expression analysis of circulating miR-21, miR-34a and miR-122 and redox status markers in metabolic Dysfunction-associated steatotic liver disease patients with and without type 2 diabetes. Int J Mol Sci. 2025; 26( 6): 2392. doi:10.3390/ijms26062392. [Google Scholar] [CrossRef]
67. Ngo V , Duennwald ML . Nrf2 and oxidative stress: a general overview of mechanisms and implications in human disease. Antioxidants. 2022; 11( 12): 2345. doi:10.3390/antiox11122345. [Google Scholar] [CrossRef]
68. Smirne C , Croce E , Di Benedetto D , Cantaluppi V , Comi C , Sainaghi PP , et al. Oxidative stress in non-alcoholic fatty liver disease. Livers. 2022; 2( 1): 30– 76. doi:10.3390/livers2010003. [Google Scholar] [CrossRef]
69. Li C , Deng D , Jiang Q , Shi J , Xu L , Liu Y . Ferroptosis in NAFLD: insights and the therapeutic potential of exercise. Front Med. 2025; 12: 1462145. doi:10.3389/fmed.2025.1462145. [Google Scholar] [CrossRef]
70. Raza S , Tewari A , Rajak S , Sinha RA . Vitamins and non-alcoholic fatty liver disease: a Molecular Insight. Liver Res. 2021; 5( 2): 62– 71. doi:10.1016/j.livres.2021.03.004. [Google Scholar] [CrossRef]
71. Kulkarni SR , Donepudi AC , Xu J , Wei W , Cheng QC , Driscoll MV , et al. Fasting induces nuclear factor E2-related factor 2 and ATP-binding Cassette transporters via protein kinase A and Sirtuin-1 in mouse and human. Antioxid Redox Signal. 2014; 20( 1): 15– 30. doi:10.1089/ars.2012.5082. [Google Scholar] [CrossRef]
72. Mastrototaro L , Roden M . Insulin resistance and insulin sensitizing agents. Metabolism. 2021; 125: 154892. doi:10.1016/j.metabol.2021.154892. [Google Scholar] [CrossRef]
73. von Loeffelholz C , Coldewey SM , Birkenfeld AL . A narrative review on the role of AMPK on de novo lipogenesis in non-alcoholic fatty liver disease: evidence from human studies. Cells. 2021; 10( 7): 1822. doi:10.3390/cells10071822. [Google Scholar] [CrossRef]
74. Ngo HKC , Kim DH , Cha YN , Na HK , Surh YJ . Nrf2 mutagenic activation drives hepatocarcinogenesis. Cancer Res. 2017; 77( 18): 4797– 808. doi:10.1158/0008-5472.CAN-16-3538. [Google Scholar] [CrossRef]
75. Diesinger T , Lautwein A , Bergler S , Buckert D , Renz C , Dvorsky R , et al. A new CYP2E1 inhibitor, 12-imidazolyl-1-dodecanol, represents a potential treatment for hepatocellular carcinoma. Can J Gastroenterol Hepatol. 2021; 2021: 8854432. doi:10.1155/2021/8854432. [Google Scholar] [CrossRef]
76. Gan L , Wang W , Jiang J , Tian K , Liu W , Cao Z . Dual role of Nrf2 signaling in hepatocellular carcinoma: promoting development, immune evasion, and therapeutic challenges. Front Immunol. 2024; 15: 1429836. doi:10.3389/fimmu.2024.1429836. [Google Scholar] [CrossRef]
77. Peleman C , Hellemans S , Veeckmans G , Arras W , Zheng H , Koeken I , et al. Ferroptosis is a targetable detrimental factor in metabolic dysfunction-associated steatotic liver disease. Cell Death Differ. 2024; 31( 9): 1113– 26. doi:10.1038/s41418-024-01348-9. [Google Scholar] [CrossRef]
78. Mercer JR , Yu E , Figg N , Cheng KK , Prime TA , Griffin JL , et al. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/−/ApoE−/− mice. Free Radic Biol Med. 2012; 52( 5): 841– 9. doi:10.1016/j.freeradbiomed.2011.11.026. [Google Scholar] [CrossRef]
79. Lonardo A , Weiskirchen R . PPARs in molecular pathogenesis and drug treatment of type 2 diabetes-related MASLD. Explor Dig Dis. 2025; 4: 100590. doi:10.37349/edd.2025.100590. [Google Scholar] [CrossRef]
80. Zhou M , Lv J , Chen X , Shi Y , Chao G , Zhang S . From gut to liver: exploring the crosstalk between gut-liver axis and oxidative stress in metabolic dysfunction-associated steatotic liver disease. Ann Hepatol. 2025; 30( 1): 101777. doi:10.1016/j.aohep.2025.101777. [Google Scholar] [CrossRef]
81. Herrera-Ojeda JL , Blanco-Palma RS , Chávez-Tapia NC , Uribe M , Montalvo-Javé EE , Nuño-Lámbarri N . The pathophysiological link between type 1 diabetes and MASLD: insights into insulin resistance and liver dysfunction. J Endocrinol Investig. 2025; 48( 10): 2279– 93. doi:10.1007/s40618-025-02621-5. [Google Scholar] [CrossRef]
82. Shaw JF , Greatorex RA . Drugs affecting the prostaglandin synthetic pathway and rat heart allograft survival. Adv Prostaglandin Thromboxane Leukot Res. 1985; 13: 219– 20. [Google Scholar]
83. Gaul S , Leszczynska A , Alegre F , Kaufmann B , Johnson CD , Adams LA , et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol. 2021; 74( 1): 156– 67. doi:10.1016/j.jhep.2020.07.041. [Google Scholar] [CrossRef]
84. Zhao H , Zhang Y , Zhang Y , Chen C , Liu H , Yang Y , et al. The role of NLRP3 inflammasome in hepatocellular carcinoma. Front Pharmacol. 2023; 14: 1150325. doi:10.3389/fphar.2023.1150325. [Google Scholar] [CrossRef]
85. Cohen NR , Garg S , Brenner MB . Antigen presentation by CD1 lipids, T cells, and NKT cells in microbial immunity. Adv Immunol. 2009; 102: 1– 94. doi:10.1016/S0065-2776(09)01201-2. [Google Scholar] [CrossRef]
86. Santos Marques H , de Brito BB , da Silva FAF , Santos MLC , de Souza JCB , Correia TML , et al. Relationship between Th17 immune response and cancer. World J Clin Oncol. 2021; 12( 10): 845– 67. doi:10.5306/wjco.v12.i10.845. [Google Scholar] [CrossRef]
87. Wang H , Zhang H , Wang Y , Brown ZJ , Xia Y , Huang Z , et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol. 2021; 75( 6): 1271– 83. doi:10.1016/j.jhep.2021.07.032. [Google Scholar] [CrossRef]
88. Cai X , Guillot A , Liu H . Cellular senescence in hepatocellular carcinoma: the passenger or the driver? Cells. 2022; 12( 1): 132. doi:10.3390/cells12010132. [Google Scholar] [CrossRef]
89. Lokau J , Schoeder V , Haybaeck J , Garbers C . Jak-stat signaling induced by interleukin-6 family cytokines in hepatocellular carcinoma. Cancers. 2019; 11( 11): 1704. doi:10.3390/cancers11111704. [Google Scholar] [CrossRef]
90. Pogribny IP , Starlard-Davenport A , Tryndyak VP , Han T , Ross SA , Rusyn I , et al. Difference in expression of hepatic microRNAs miR-29c, miR-34a, miR-155, and miR-200b is associated with strain-specific susceptibility to dietary nonalcoholic steatohepatitis in mice. Lab Investig. 2010; 90( 10): 1437– 46. doi:10.1038/labinvest.2010.113. [Google Scholar] [CrossRef]
91. Weiskirchen R , Lonardo A . How ‘miracle’ weight-loss semaglutide promises to change medicine but can we afford the expense? Br J Pharmacol. 2025; 182( 8): 1651– 70. doi:10.1111/bph.70003. [Google Scholar] [CrossRef]
92. Lonardo A , Weiskirchen R . Tirzepatide in metabolic dysfunction-associated steatotic liver disease and steatohepatitis: a novel star on the horizon? Explor Drug Sci. 2025; 3: 1008128. doi:10.37349/eds.2025.1008128. [Google Scholar] [CrossRef]
93. Odanga JJ , Anderson SM , LeCluyse EL , Presnell SC , Chen J , Weaver JR . Effects of obeticholic acid treatment on primary human hepatocytes in a novel tri-culture model system. Cells. 2025; 14( 13): 968. doi:10.3390/cells14130968. [Google Scholar] [CrossRef]
94. Shao N , Qiu H , Liu J , Xiao D , Zhao J , Chen C , et al. Targeting lipid metabolism of macrophages: a new strategy for tumor therapy. J Adv Res. 2025; 68: 99– 114. doi:10.1016/j.jare.2024.02.009. [Google Scholar] [CrossRef]
95. Groth C , Weber R , Lasser S , Özbay FG , Kurzay A , Petrova V , et al. Tumor promoting capacity of polymorphonuclear myeloid-derived suppressor cells and their neutralization. Int J Cancer. 2021; 149( 9): 1628– 38. doi:10.1002/ijc.33731. [Google Scholar] [CrossRef]
96. Koo SY , Park EJ , Lee CW . Immunological distinctions between nonalcoholic steatohepatitis and hepatocellular carcinoma. Exp Mol Med. 2020; 52( 8): 1209– 19. doi:10.1038/s12276-020-0480-3. [Google Scholar] [CrossRef]
97. Wang F , Zhang X , Liu W , Zhou Y , Wei W , Liu D , et al. Activated natural killer cell promotes nonalcoholic steatohepatitis through mediating JAK/STAT pathway. Cell Mol Gastroenterol Hepatol. 2022; 13( 1): 257– 74. doi:10.1016/j.jcmgh.2021.08.019. [Google Scholar] [CrossRef]
98. Ge Z , Zhou G , Campos Carrascosa L , Gausvik E , Boor PPC , Noordam L , et al. TIGIT and PD1 co-blockade Restores ex vivo Functions of Human Tumor-Infiltrating CD8+ T Cells in Hepatocellular Carcinoma. Cell Mol Gastroenterol Hepatol. 2021; 12( 2): 443– 64. doi:10.1016/j.jcmgh.2021.03.003. [Google Scholar] [CrossRef]
99. Supruniuk E , Grubczak K , Parfieniuk-Kowerda A , Flisiak R , Moniuszko M , Jaroszewicz J , et al. Increased frequencies of human Th-17 CD4+ T-cells and decreased T-regulatory cells in patients with early and advanced metabolic dysfunction-associated steatotic liver disease. Front Immunol. 2025; 16: 1597204. doi:10.3389/fimmu.2025.1597204. [Google Scholar] [CrossRef]
100. Patseas D , El-Masry A , Liu Z , Ramachandran P , Triantafyllou E . Myeloid cells in chronic liver inflammation. Cell Mol Immunol. 2025; 22( 10): 1237– 61. doi:10.1038/s41423-025-01324-4. [Google Scholar] [CrossRef]
101. Jiang Z , Qian B , Xu T , Bai J , Fu W . Immune microenvironment on the molecular mechanisms and therapeutic targets of MAFLD. Immunotargets Ther. 2025; 14: 719– 33. doi:10.2147/ITT.S530451. [Google Scholar] [CrossRef]
102. Riaz F , Wei P , Pan F . Fine-tuning of regulatory T cells is indispensable for the metabolic steatosis-related hepatocellular carcinoma: a review. Front Cell Dev Biol. 2022; 10: 949603. doi:10.3389/fcell.2022.949603. [Google Scholar] [CrossRef]
103. Ramos-Tovar E , Muriel P . NLRP3 inflammasome in hepatic diseases: a pharmacological target. Biochem Pharmacol. 2023; 217: 115861. doi:10.1016/j.bcp.2023.115861. [Google Scholar] [CrossRef]
104. Subczynski WK , Pasenkiewicz-Gierula M , Widomska J , Mainali L , Raguz M . High cholesterol/low cholesterol: effects in biological membranes: a review. Cell Biochem Biophys. 2017; 75( 3–4): 369– 85. doi:10.1007/s12013-017-0792-7. [Google Scholar] [CrossRef]
105. Vizioli G , Nicoletti A , Feliciani D , Funaro B , Zileri Dal Verme L , Ponziani FR , et al. Immunotherapy and MASLD-related HCC: should we reconsider the role of etiology in the therapeutic approach to HCC? Appl Sci. 2025; 15( 5): 2279. doi:10.3390/app15052279. [Google Scholar] [CrossRef]
106. Henry Z , Meadows V , Guo GL . FXR and NASH: an avenue for tissue-specific regulation. Hepatol Commun. 2023; 7( 5): e0127. doi:10.1097/HC9.0000000000000127. [Google Scholar] [CrossRef]
107. Wree A , McGeough MD , Inzaugarat ME , Eguchi A , Schuster S , Johnson CD , et al. NLRP3 inflammasome driven liver injury and fibrosis: roles of IL-17 and TNF in mice. Hepatology. 2018; 67( 2): 736– 49. doi:10.1002/hep.29523. [Google Scholar] [CrossRef]
108. Han J , Wang Y . mTORC1 signaling in hepatic lipid metabolism. Protein Cell. 2018; 9( 2): 145– 51. doi:10.1007/s13238-017-0409-3. [Google Scholar] [CrossRef]
109. Dodd KM , Yang J , Shen MH , Sampson JR , Tee AR . mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene. 2015; 34( 17): 2239– 50. doi:10.1038/onc.2014.164. [Google Scholar] [CrossRef]
110. Kawauchi K , Araki K , Tobiume K , Tanaka N . p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008; 10( 5): 611– 8. doi:10.1038/ncb1724. [Google Scholar] [CrossRef]
111. An H , Jang Y , Choi J , Hur J , Kim S , Kwon Y . New insights into AMPK, as a potential therapeutic target in metabolic dysfunction-associated steatotic liver disease and hepatic fibrosis. Biomol Ther. 2025; 33( 1): 18– 38. doi:10.4062/biomolther.2024.188. [Google Scholar] [CrossRef]
112. Baker RG , Hayden MS , Ghosh S . NF-κB, inflammation, and metabolic disease. Cell Metab. 2011; 13( 1): 11– 22. doi:10.1016/j.cmet.2010.12.008. [Google Scholar] [CrossRef]
113. Liu X , Chhipa RR , Pooya S , Wortman M , Yachyshin S , Chow LML , et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci U S A. 2014; 111( 4): E435– 44. doi:10.1073/pnas.1311121111. [Google Scholar] [CrossRef]
114. Goldberg EM , Zidovetzki R . Synergistic effects of diacylglycerols and fatty acids on membrane structure and protein kinase C activity. Biochemistry. 1998; 37( 16): 5623– 32. doi:10.1021/bi9719354. [Google Scholar] [CrossRef]
115. Eski SE , Mi J , Pozo-Morales M , Hovhannisyan GG , Perazzolo C , Manco R , et al. Cholangiocytes contribute to hepatocyte regeneration after partial liver injury during growth spurt in zebrafish. Nat Commun. 2025; 16( 1): 5260. doi:10.1038/s41467-025-60334-y. [Google Scholar] [CrossRef]
116. Aoshima K , Tanimizu N . Lineage plasticity and reprogramming of epithelial cells during tissue injury and regeneration-lessons from the lineage plasticity of hepatocytes and cholangiocytes induced by liver injury. Regen Ther. 2025; 29: 447– 54. doi:10.1016/j.reth.2025.04.008. [Google Scholar] [CrossRef]
117. Vancura A , Nagar S , Kaur P , Bu P , Bhagwat M , Vancurova I . Reciprocal regulation of AMPK/SNF1 and protein acetylation. Int J Mol Sci. 2018; 19( 11): 3314. doi:10.3390/ijms19113314. [Google Scholar] [CrossRef]
118. Wu Y , Deng J , Rychahou PG , Qiu S , Evers BM , Zhou BP . Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009; 15( 5): 416– 28. doi:10.1016/j.ccr.2009.03.016. [Google Scholar] [CrossRef]
119. Wu X , Xu Y , Liang Q , Yang X , Huang J , Wang J , et al. Recent advances in dual PI3K/mTOR inhibitors for tumour treatment. Front Pharmacol. 2022; 13: 875372. doi:10.3389/fphar.2022.875372. [Google Scholar] [CrossRef]
120. Qian S , Rui M , Kutbi E , AlJurayyan AN , Abu-Zaid A , Jamilian P , et al. Imeglimin as an effective therapeutic approach in management of type 2 diabetes mellitus: an umbrella review and systematic review, meta-regression and meta-analysis. Diabetol Metab Syndr. 2025; 17( 1): 357. doi:10.1186/s13098-025-01922-2. [Google Scholar] [CrossRef]
121. Doupis J , Baris N , Avramidis K . Imeglimin: a new promising and effective weapon in the treatment of type 2 diabetes. touchREV Endocrinol. 2021; 17( 2): 88– 91. doi:10.17925/EE.2021.17.2.88. [Google Scholar] [CrossRef]
122. Prescott JA , Cook SJ . Targeting IKKβ in cancer: challenges and opportunities for the therapeutic utilisation of IKKβ inhibitors. Cells. 2018; 7( 9): 115. doi:10.3390/cells7090115. [Google Scholar] [CrossRef]
123. Kietzmann T . Metabolic zonation of the liver: the oxygen gradient revisited. Redox Biol. 2017; 11: 622– 30. doi:10.1016/j.redox.2017.01.012. [Google Scholar] [CrossRef]
124. Iwakiri Y , Shah V , Rockey DC . Vascular pathobiology in chronic liver disease and cirrhosis—current status and future directions. J Hepatol. 2014; 61( 4): 912– 24. doi:10.1016/j.jhep.2014.05.047. [Google Scholar] [CrossRef]
125. Chen JA , Shi M , Li JQ , Qian CN . Angiogenesis: multiple masks in hepatocellular carcinoma and liver regeneration. Hepatol Int. 2010; 4( 3): 537– 47. doi:10.1007/s12072-010-9192-4. [Google Scholar] [CrossRef]
126. Lurje I , Gaisa NT , Weiskirchen R , Tacke F . Mechanisms of organ fibrosis: emerging concepts and implications for novel treatment strategies. Mol Aspects Med. 2023; 92: 101191. doi:10.1016/j.mam.2023.101191. [Google Scholar] [CrossRef]
127. Felli E , Nulan Y , Ortega-Ribera M , Fernández-Iglesias A , Gracia-Sancho J . The role of liver sinusoidal endothelial cells in liver diseases: key players in health and pathology. J Hepatol. 2025. doi:10.1016/j.jhep.2025.10.017. [Google Scholar] [CrossRef]
128. Siemann DW . The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat Rev. 2011; 37( 1): 63– 74. doi:10.1016/j.ctrv.2010.05.001. [Google Scholar] [CrossRef]
129. Hirayama AB , Taliberti IB , de Oliveira CPMS , Alves VAF . Lipotoxicity plays a key role in the development of angiogenesis and microcirculatory modulation in masld spectrum. Arq Gastroenterol. 2025; 62: e25053. doi:10.1590/S0004-2803.24612025-053. [Google Scholar] [CrossRef]
130. Finn RS , Qin S , Ikeda M , Galle PR , Ducreux M , Kim TY , et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020; 382( 20): 1894– 905. doi:10.1056/NEJMoa1915745. [Google Scholar] [CrossRef]
131. Dekervel J , Casadei-Gardini A , Pinter M . Controversies in upper GI oncology: first-line systemic treatment in MASLD-associated HCC. ESMO Open. 2024; 9( 2): 102245. doi:10.1016/j.esmoop.2024.102245. [Google Scholar] [CrossRef]
132. Wang Z , Sheng J , Zhang X . Characterization of adverse reactions to four common targeted drugs for hepatocellular carcinoma in WHO-VigiAccess. Sci Rep. 2025; 15( 1): 16188. doi:10.1038/s41598-025-00004-7. [Google Scholar] [CrossRef]
133. Zargar AH , Bhansali A , Majumdar A , Maheshwari A , Bhattacharyya A , Dasgupta A , et al. Management of metabolic dysfunction-associated steatotic liver disease (MASLD)—an expert consensus statement from Indian diabetologists’ perspective. Diabetes Obes Metab. 2025; 27( Suppl 4): 3– 20. doi:10.1111/dom.16496. [Google Scholar] [CrossRef]
134. Ma Y , Wang J , Xiao W , Fan X . A review of MASLD-related hepatocellular carcinoma: progress in pathogenesis, early detection, and therapeutic interventions. Front Med. 2024; 11: 1410668. doi:10.3389/fmed.2024.1410668. [Google Scholar] [CrossRef]
135. Vitellius C , Desjonqueres E , Lequoy M , Amaddeo G , Fouchard I , N’Kontchou G , et al. MASLD-related HCC: multicenter study comparing patients with and without cirrhosis. JHEP Rep. 2024; 6( 10): 101160. doi:10.1016/j.jhepr.2024.101160. [Google Scholar] [CrossRef]
136. Vitale A , Angelico R , Sensi B , Lai Q , Kauffmann E , Scalera I , et al. What is the role of minimally invasive liver surgery in treating patients with hepatocellular carcinoma on cirrhosis? Cancers. 2024; 16( 5): 966. doi:10.3390/cancers16050966. [Google Scholar] [CrossRef]
137. Norris P , Gow J , Arthur T , Conway A , Fleming FJ , Ralph N . Metabolic syndrome and surgical complications: a systematic review and meta-analysis of 13 million individuals. Int J Surg. 2024; 110( 1): 541– 53. doi:10.1097/JS9.0000000000000834. [Google Scholar] [CrossRef]
138. Henry ZH , Argo CK . Management of chronic liver disease in patients with hepatocellular carcinoma. Clin Liver Dis. 2025; 29( 1): 135– 47. doi:10.1016/j.cld.2024.08.004. [Google Scholar] [CrossRef]
139. Delacôte C , Favre M , El Amrani M , Ningarhari M , Lemaitre E , Ntandja-Wandji LC , et al. Morbid obesity increases death and dropout from the liver transplantation waiting list: a prospective cohort study. United Eur Gastroenterol J. 2022; 10( 4): 396– 408. doi:10.1002/ueg2.12226. [Google Scholar] [CrossRef]
140. Feng G , Han Y , Yang W , Shikora S , Mahawar K , Cheung TT , et al. Recompensation in MASLD-related cirrhosis via metabolic bariatric surgery. Trends Endocrinol Metab. 2025; 36( 2): 118– 32. doi:10.1016/j.tem.2024.05.009. [Google Scholar] [CrossRef]
141. García-Sesma A , Calvo J , Manrique A , Cambra F , Justo I , Caso O , et al. Morbidly obese patients awaiting liver transplantation-sleeve gastrectomy: safety and efficacy from a liver transplant unit experience. Transplant Proc. 2019; 51( 1): 33– 7. doi:10.1016/j.transproceed.2018.01.060. [Google Scholar] [CrossRef]
142. Provera A , Vecchio C , Sheferaw AN , Stoppa I , Pantham D , Dianzani U , et al. From MASLD to HCC: what’s in the middle? Heliyon. 2024; 10( 15): e35338. doi:10.1016/j.heliyon.2024.e35338. [Google Scholar] [CrossRef]
143. Lonardo A . Principles of risk stratification in nonalcoholic fatty liver disease. A narrative review emphasizing non-invasive strategies. Explor Dig Dis. 2023: 188– 201. doi:10.37349/edd.2023.00026. [Google Scholar] [CrossRef]
144. Burke L , Hinkson A , Haghnejad V , Jones R , Parker R , Rowe IA . Hepatocellular carcinoma risk scores for non-viral liver disease: a systematic review and meta-analysis. JHEP Rep. 2024; 7( 1): 101227. doi:10.1016/j.jhepr.2024.101227. [Google Scholar] [CrossRef]
145. Lai JC , Yang B , Lee HW , Lin H , Tsochatzis EA , Petta S , et al. Non-invasive risk-based surveillance of hepatocellular carcinoma in patients with metabolic dysfunction-associated steatotic liver disease. Gut. 2025; 74( 12): 2050– 7. doi:10.1136/gutjnl-2025-334981. [Google Scholar] [CrossRef]
146. Liu M , Liu C , Mok TN , Qi X , Ming WK . Research communication: changing aetiology of chronic liver diseases in east Asia Pacific and HCC surveillance in non-cirrhotic patients. Aliment Pharmacol Ther. 2025. doi:10.1111/apt.70428. [Google Scholar] [CrossRef]
147. Harrison SA , Bedossa P , Guy CD , Schattenberg JM , Loomba R , Taub R , et al. A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. N Engl J Med. 2024; 390( 6): 497– 509. doi:10.1056/nejmoa2309000. [Google Scholar] [CrossRef]
148. Sanyal AJ , Newsome PN , Kliers I , Østergaard LH , Long MT , Kjær MS , et al. Phase 3 trial of semaglutide in metabolic dysfunction–associated steatohepatitis. N Engl J Med. 2025; 392( 21): 2089– 99. doi:10.1056/nejmoa2413258. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools