
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Neuroinflammation and Oxidative Stress: Their Pathophysiological Roles in Amyotrophic Lateral Sclerosis and Alzheimer’s Disease
1 Department of Biophysics, Near East University, Nicosia, North Cyprus via Mersin 10, Türkiye
2 Department of Biochemistry, Faculty of Pharmacy, Eastern Mediterranean University, Famagusta, North Cyprus via Mersin 10, Türkiye
3 Department of Pharmacology, Faculty of Dentistry, Near East University, Nicosia, North Cyprus via Mersin 10, Türkiye
* Corresponding Authors: Aslı Aykaç. Email: ,
(This article belongs to the Special Issue: Cellular and Molecular Mechanisms of Neurodegeneration: From Pathogenesis to Therapeutic Strategies)
BIOCELL 2026, 50(7), 4 https://doi.org/10.32604/biocell.2026.077114
Received 02 December 2025; Accepted 10 February 2026; Issue published 29 June 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Neurodegenerative diseases such as Amyotrophic Lateral Sclerosis (ALS) and Alzheimer’s disease (AD) are driven by complex, multifactorial mechanisms in which oxidative stress (OS) and neuroinflammation (NI) play central, mutually reinforcing roles. Their interaction is mediated through key signaling pathways, including nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), nuclear factor erythroid 2–related factor 2–Kelch-like ECH-associated protein 1 (Nrf2-Keap1), and the mitogen-activated protein kinase (MAPK) pathway, as well as mitochondrial dysfunction, microglial activation, and dysregulated redox homeostasis. Increasing attention has been directed toward understanding how Food and Drug Administration (FDA)-approved neuroprotective agents influence these interconnected processes. Drugs such as riluzole, edaravone, memantine, and acetylcholinesterase inhibitors exhibit diverse effects on glutamate excitotoxicity, free radical production, inflammatory cytokine release, and microglial reactivity, yet their modest clinical efficacy highlights the limitations of single-target approaches. This review synthesizes current evidence on the mechanistic relationship between OS and NI in ALS and AD and evaluates how existing therapeutics modulate these pathways, emphasizing the need for multitarget strategies to achieve meaningful disease-modifying outcomes.Graphic Abstract
Keywords
Neurodegenerative diseases represent a series of progressive and often irreversible conditions that lead to devastating neuronal loss. The hallmark clinical symptoms of these diseases, including Amyotrophic Lateral Sclerosis (ALS) and Alzheimer’s disease (AD), manifest as a gradual decline in essential motor and cognitive functions, significantly impairing the quality of life over time. The mechanisms driving these complex diseases are multifaceted and involve multiple contributing factors [1,2]. While genetic predisposition, proteostatic disorders, and environmental influences have long been recognized, emerging research underscores the critical roles of neuroinflammation (NI) and oxidative stress (OS). These two factors are now understood to be pivotal in triggering and perpetuating the progression of neurodegenerative diseases, amplifying the urgency for targeted interventions and innovative therapeutic strategies [3,4,5]. Both experimental and clinical research results indicate that the interaction between OS and NI causes permanent damage to nerve cells and plays a decisive role in disease progression [4,6].
The treatment of neurodegenerative diseases is not only symptomatic but also targets neuroprotective agents that address underlying processes, such as OS and NI [2]. Evaluating the efficacy of FDA-approved drugs for neurodegenerative diseases in processes such as NI and OS is a focal point of translational neuroscience research today [7,8,9]. This review aims to elucidate the mechanistic interplay between oxidative stress and neuroinflammation in the pathogenesis of ALS and AD, and to evaluate how FDA-approved therapeutics modulate these interconnected pathways. The paper contributes to the field by integrating mitochondrial biology, redox signaling, inflammasome activation, and glial immunometabolism into a unified framework that explains how oxidative and inflammatory processes co-evolve during neurodegeneration. The novelty of this work lies in its comprehensive synthesis of matrix-derived Reactive Oxygen Species (ROS) dynamics and disease-specific mitochondrial vulnerabilities in ALS and AD, offering a mechanistic rationale for repurposing existing therapeutics and guiding the development of future combination strategies that simultaneously target oxidative stress and neuroinflammation.
A narrative review was conducted to examine the interplay between oxidative stress and neuroinflammation in the pathophysiological mechanisms underlying ALS and AD, and to evaluate the effects of FDA-approved medications on these processes. Literature searches were performed using Google Scholar, PubMed, and Web of Science. The search strategy combined the following keywords: “Alzheimer’s disease”, “Amyotrophic Lateral Sclerosis”, “neuroinflammation”, “oxidative stress”, and “FDA-approved drugs.” Articles published between 1994 and 2025 were considered. Studies focusing on diseases other than ALS and AD were excluded from the review.
3 The Cellular Basis of Neuroinflammation
According to Berríos-Cárcamo et al. (2020), the brain, an organ with highly specialized immune surveillance, is largely protected from peripheral immune cells and circulating signals by the blood–brain barrier (BBB) [10]. Despite this relative isolation, the central nervous system (CNS) can mount an inflammatory response to local pathogens or tissue injury. This response is primarily mediated by astrocytes, the key regulators of neuronal and metabolic homeostasis, and microglia, the resident immune cells of the brain. In the CNS, oligodendrocytes, microglia, and/or astrocytes release proinflammatory mediators (cytokines, chemokines, etc.), which initiate and prolong the neuroinflammatory response [3,11].
However, the brain can mount its own inflammatory response to local pathogens or tissue injury through the activation of astrocytes, the primary regulators of neuronal, metabolic, and synaptic homeostasis, and microglia, the resident immune cells of the CNS. Both cell types produce cytokines and express pattern recognition receptors, including Toll-like receptors (TLRs), which enable them to monitor the microenvironment and initiate appropriate inflammatory signaling. Astrocytes play a central role in shaping the neuroinflammatory response. Upon exposure to danger-associated or pathogen-associated signals, astrocytes undergo reactive astrogliosis, characterized by hypertrophy, upregulation of glial fibrillary acidic protein (GFAP), and transcriptional reprogramming. Activated astrocytes release pro-inflammatory mediators such as Interleukin-1 beta (IL-1β), Tumor necrosis factor-alpha (TNF-α), and IL-6, modulate chemokine gradients that recruit or influence microglial activity, and regulate extracellular glutamate levels through altered expression of glutamate transporters (e.g., EAAT1/EAAT2), thereby contributing to excitotoxic stress [12,13]. They also influence oxidative stress by altering antioxidant defenses, including glutathione metabolism, and by producing nitric oxide and other reactive species under sustained inflammatory conditions. Through these mechanisms, astrocytes amplify or sustain neuroinflammation and propagate neuronal dysfunction. In response to pro-inflammatory stimuli, microglia undergo phenotypic and transcriptional changes, leading to the upregulation of oxidant-generating enzymes such as NADPH oxidase (NOX) and inducible nitric oxide synthase (iNOS). These enzymes drive the production of ROS and reactive nitrogen species (RNS), further intensifying oxidative stress within the neural milieu [14].
Microglial cells normally secrete neurotrophic factors to maintain tissue homeostasis. Still, when overstimulated, they switch to a proinflammatory phenotype, increasing the release of various cytokines [TNF-α, IL-1β, IL-6, etc.] (Fig. 1) [3,15,16]. The resulting proinflammatory environment triggers the activation of the apoptotic pathway in neurons, primarily through Ca2+ imbalance and mitochondrial membrane damage. Long-term microglial activation causes glial cells to enter a state called “priming”, leading to excessive responses even to mild stimuli. This pattern is frequently observed in patients with AD and ALS [17,18]. The proinflammatory environment created by microglial activation also triggers OS by increasing ROS production. For this very reason, NI is considered not only a defense response but also a cyclical mechanism that perpetuates neuronal toxicity [19,20].

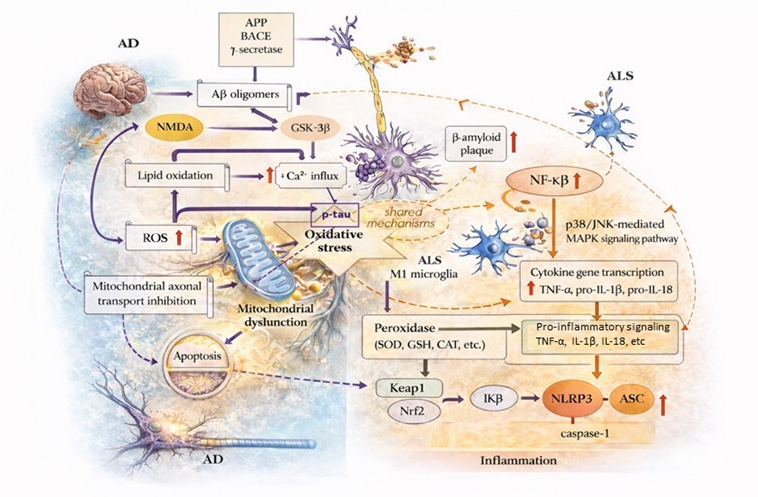
Figure 1: Common and disease-specific pathophysiological mechanisms in Alzheimer’s disease (AD) and Amyotrophic Lateral Sclerosis (ALS). This figure is adapted from Iliyasu et al. (2023) [21]. This figure summarizes the key molecular and cellular pathways involved in the pathogenesis of AD and ALS, as well as the common pathophysiological intersections among them. In AD, Aβ oligomers formed by β-secretase (BACE) and γ-secretase-mediated proteolytic processing of amyloid precursor protein (APP) trigger neuronal dysfunction via N-methyl-D-aspartate (NMDA) receptor activation and glycogen synthase kinase-3 beta (GSK-3β) signaling pathways. GSK-3β activation initiates two parallel and independent pathological processes: excitotoxicity, characterized by increased Ca2+ influx, and hyperphosphorylation of tau proteins (p-tau). p-tau accumulation disrupts microtubule dynamics and inhibits mitochondrial axonal transport, weakening neuronal integrity and indirectly contributing to cellular death. Aβ-mediated lipid oxidation and increased production of reactive oxygen species (ROS) lead to mitochondrial dysfunction and oxidative stress. Oxidative stress potentiates inflammatory responses and microglial activation as a central pathogenic axis shared in AD and ALS. In the context of ALS, activation of microglia in the M1 phenotype results in increased p38/JNK-mediated MAPK signaling and nuclear factor kappa B (NF-κB) activity, stimulating transcription of pro-inflammatory cytokine genes (TNF-α, pro-IL-1β, pro-IL-18). This process, driven by the activation of the NLRP3 inflammasome complex (NLRP3, ASC, and caspase-1), sustains chronic neuroinflammation. Antioxidant defense mechanisms are regulated via the Keap1–Nrf2 axis, which controls superoxide dismutase (SOD), catalase (CAT), glutathione (GSH), and related peroxidase systems. Disruption of these protective mechanisms contributes to increased oxidative stress and inflammatory signaling. Overall, the diagram illustrates the mutually reinforcing pathological cycles of oxidative stress, mitochondrial dysfunction, and inflammation in AD and ALS, accelerating disease progression. The yellow upward arrow represented an increase. APP, Amyloid precursor protein; Aβ, Beta-amyloid; BACE, β-secretase; ROS, Reactive oxygen species; NMDA, N-methyl-D-aspartate; GSK-3β, Glycogen synthase kinase 3 beta; p-tau, Phosphorylated tau; ALS, Amyotrophic lateral sclerosis; TNF-α, Tumor necrosis factor-α; IL-18, Interleukin-18; IL-1β, Interleukin-1β; GSH, Glutathione; NF-κB, Nuclear factor kappa B; SOD, Superoxide Dismutase; CAT, Catalase; NLRP3, NLR family pyrin domain containing-3; ASC, apoptotic speck-like protein containing a caspase recruitment domain; Nrf2, Nuclear Factor Erythroid 2-Related Factor 2; Keap 1, Kelch-like ECH-associated protein 1; Ca2+, Calcium.
4 Molecular Dynamics of Oxidative Stress
OS and NI are driven by tightly coupled molecular events within neural tissue, with mitochondria serving as the principal source of ROS. Electron leakage from Complexes I–III of the electron transport chain generates superoxide, but Complex I is the dominant source of ROS within the mitochondrial matrix, where oxidative damage is particularly harmful. Matrix-directed ROS formation arises through two mechanistically distinct processes. During forward electron transfer (FET), electrons flow from NADH to ubiquinone; a fraction of them prematurely reduce oxygen at the FMN site, generating superoxide directly in the matrix. Under metabolic stress, ischemia-reperfusion, or impaired downstream electron flow, reverse electron transfer (RET) occurs, in which electrons move backward from ubiquinol to Complex I. RET is one of the most potent physiological triggers of ROS production, generating orders of magnitude more superoxide than FET due to the combination of a highly reduced ubiquinone pool and an elevated mitochondrial membrane potential [22]. The matrix environment—rich in NADH, flavin adenine mononucleotide (FMNH2), iron–sulfur clusters, and tightly packed respiratory complexes—further favors one-electron oxygen reduction. Because ROS are produced adjacent to mitochondrial DNA, cardiolipin-rich inner membranes, and TCA cycle enzymes, matrix ROS inflict disproportionate structural and metabolic damage [23].
Accumulation of ROS and RNS disrupts redox homeostasis, initiating lipid peroxidation, DNA breaks, and protein oxidation [24]. These oxidative insults activate redox-sensitive inflammatory pathways, particularly NF-κβ, which induces IL-1β, Cyclooxygenase-2 (COX-2), TNF-α, and iNOS, thereby amplifying microglial and astrocytic activation. Activated glia further increase ROS production through NOX enzymes and nitric oxide pathways, reinforcing mitochondrial dysfunction [25,26]. In contrast, the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway coordinates antioxidant defense by upregulating heme oxygenase-1, glutathione peroxidase, and other detoxifying enzymes; however, chronic inflammation and aging diminish Nrf2 activity, shifting the balance toward sustained NF-κB activation. This reciprocal suppression of antioxidant defenses and reinforcement of inflammatory signaling establishes a self-perpetuating cycle in which oxidative stress and neuroinflammation mutually intensify, driving progressive neuronal vulnerability [27,28].
5 The Bidirectional Microglia–Mitochondria Interaction
It has been reported that dysregulation of inflammatory responses triggered by mitochondrial components or products contributes to numerous human diseases, ranging from those driven by excessive inflammation to those arising from inefficient inflammatory reactions [29]. In recent years, it has been shown that mitochondrial function is not limited to energy production but also plays an active role in regulating the immune response [25,26]. During microglial activation, elevated levels of mitochondria-derived reactive oxygen species (mtROS) serve as key triggers for the assembly of NLRP3 inflammasome complexes, leading to caspase-1 activation and the subsequent release of IL-1β and IL-18, which amplify the inflammatory response. Multiple signaling pathways link mitochondrial dysfunction to innate immune activation. One major pathway involves the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS), which is activated when damaged mitochondria release oxidized mitochondrial DNA (mtDNA) into the cytosol. Activated cGAS produces cyclic GMP-AMP (cGAMP), which binds to and activates the adaptor protein interferon response cGAMP interactor 1 (STING1), initiating type I interferon signaling and promoting a pro-inflammatory transcriptional program. In parallel, both mtROS and cytosolic mtDNA directly stimulate inflammasome activation, particularly NLR family pyrin domain containing 3 (NLRP3), by promoting thiol oxidation, mitochondrial membrane destabilization, and the release of mitochondrial danger-associated molecular patterns. Together, these pathways illustrate how mitochondrial dysfunction serves as a central molecular hub, integrating mtROS, mtDNA leakage, and innate immune signaling to drive sustained neuroinflammation [30,31,32].
Mitochondrial ROS activate inflammasome complexes (NLRP3), leading to the release of IL-1β and IL-18. Conversely, inflammation can further exacerbate mitochondrial dysfunction, creating a bidirectional cycle between oxidative stress and innate immune activation. Pro-inflammatory cytokines such as TNF-α, IL-1β, and interferon gamma (IFN-γ) impair electron transport chain activity, reduce mitochondrial membrane potential, and increase mitochondrial permeability transition, all of which promote additional mitochondrial ROS production. Activated microglia and astrocytes release nitric oxide and RNS that inhibit Complex I and Complex IV, leading to electron leakage and further mitochondrial damage. Inflammatory signaling also disrupts mitochondrial dynamics by promoting excessive fission, inhibiting fusion, and impairing mitophagy, resulting in the accumulation of dysfunctional mitochondria that release even more ROS and mitochondrial DNA into the cytosol. Thus, inflammation not only arises from mitochondrial dysfunction but also feeds back to intensify it, reinforcing a self-perpetuating cycle of mitochondrial impairment, oxidative stress, and neuroinflammation [33,34]. TNF-α and IL-6 have been reported to reduce mitochondrial membrane potential and limit ATP production. Due to this bidirectional interaction, OS and inflammation are characterized as a “vicious cycle” that continuously feeds each other [35]. In both ALS and AD, mitochondrial dysfunction and the resulting release of mitochondrial danger signals act as central drivers of innate immune activation. In AD, amyloid-β (Aβ) aggregates impair Complex I activity and destabilize mitochondrial membranes, promoting excessive mitochondrial ROS (mtROS) production and leakage of oxidized mtDNA into the cytosol. Cytosolic mtDNA activates the cGAS–STING pathway, triggering type I interferon signaling and amplifying microglial inflammatory responses. Aβ also directly enhances STING activation, further reinforcing this pro-inflammatory cascade. In ALS, mutant superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TDP-43), and Fused in Sarcoma (FUS) proteins accumulate on the outer mitochondrial membrane and within the matrix, disrupting electron transport, increasing mtROS generation, and promoting mtDNA release. These mitochondrial disturbances activate both the NLRP3 inflammasome and the cGAS–STING axis, leading to caspase-1 activation and the maturation of IL-1β and IL-18. Thus, in both diseases, pathological protein aggregates converge on a shared mechanism—mtROS-driven mitochondrial damage and mtDNA leakage—that fuels chronic neuroinflammation through NLRP3 and cGAS–STING signaling [30,36].
6 Signaling Pathways of Neuroinflammation and Oxidative Stress
The interaction between NI and OS in pathophysiological processes largely depends on the activation of specific signaling pathways. These pathways encompass a broad network ranging from the regulation of intracellular stress responses to the synthesis of inflammatory mediators [6]. The Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-κB), Nrf2, Kelch-like ECH-associated protein 1 (Keap1), Mitogen-Activated Protein Kinase (MAPK), and inflammasome signaling pathways are recognized as the primary molecular mediators of neuronal damage in neurodegenerative diseases (Fig. 1) [37,38,39].
NF-κB, a key transcription factor that regulates proinflammatory gene expression, is usually found in the cytoplasm in an inactive state, bound to its inhibitory protein IκB [40]. Under OS conditions, ROS induce the phosphorylation and degradation of IκBα, leading to the nuclear translocation of NF-κB. As a result of this process, the expression of inflammatory genes such as IL-1β, TNF-α, COX-2, and iNOS increases [4,6]. Chronic NF-κB activation leads to the conversion of microglial cells to a proinflammatory phenotype and to deeper oxidative damage in surrounding tissues [41]. Post-mortem analyses in AD and PD patients indicate increased NF-κB activity, which correlates positively with the severity of neurodegeneration [42,43].
6.2 Nrf2–Keap1 Signaling Pathway
The pathway that counterbalances NF-κB is the main regulator of the antioxidant defense system. Under physiological conditions, Nrf2 is kept inactive by forming a complex with the Keap1 protein in the cytoplasm [44]. Under OS, oxidation of critical cysteine residues in Keap1 releases Nrf2, allowing it to translocate to the nucleus, where it binds to antioxidant response elements (ARE). This binding induces the expression of antioxidant enzymes, including heme oxygenase-1, SOD, and glutathione peroxidase [44]. Decreased Nrf2 activity in conditions such as aging or chronic NI leads to the accumulation of oxidative damage and the predominance of NF-κB activity [45]. This dynamic is considered an important balancing mechanism that explains the progressive nature of neurodegenerative diseases.
The MAPK family is a multi-step phosphorylation cascade that modulates cellular stress responses. Within this family, the c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38 MAPK subunits, in particular, play a critical role in neuronal cell death [46,47]. MAPK activation, triggered by ROS accumulation or inflammatory cytokines, both increases proapoptotic gene expression and contributes to the progression of the inflammatory process by enhancing NF-κB signaling [48]. In ALS animal models, excessive activation of the MAPK cascade has been shown to accelerate neuronal loss, whereas inhibitors of p38 or JNK increase neuronal survival [49,50]. These findings suggest the pathway’s potential as a therapeutic target.
Another mechanism that directly links OS and NI is the activation of the inflammasome complex. The NLRP3, in particular, is triggered by increased mitochondrial ROS. NLRP3 activation promotes caspase-1 maturation and consequently the release of potent proinflammatory cytokines (IL-1β and IL-18) [51]. Chronic inflammasome activation deepens NI damage by driving a persistent proinflammatory phenotype in microglial cells. Elevated NLRP3 activation markers in brain tissue from AD patients support the importance of this pathway in disease progression [52].
6.5 The Integrative Effect of Signaling Networks and Their Relationship with Neuroprotective Agents
The four main signaling pathways interact with each other. While excessive NF-κB activation initiates the inflammatory process, decreased Nrf2 activity weakens antioxidant defenses. The MAPK and inflammasome pathways, on the other hand, reinforce the process by acting at the intersection of these two systems. Therefore, a cyclical mechanism emerges in which NI and OS mutually trigger one another. The therapeutic effects of neuroprotective drugs are also primarily based on these molecular networks. For example, riluzole reduces ROS production by inhibiting glutamate release [53]. Edaravone supports Nrf2 activation by directly scavenging free radicals and reduces the level of 3-nitrotyrosine in CSF, a marker of oxidative damage [7,54]. The balancing effect of these agents on the NF-κB and Nrf2 axes is considered an essential strategic approach in breaking the NI-OS cycle.
7 Cross-Mechanisms: Neuroinflammation–Oxidative Stress Interaction
Current data suggests another vicious cycle. OS not only triggers inflammation, but inflammation also increases ROS production [55]. In fact, OS and NI can be defined as two processes that mutually reinforce each other in neurodegenerative diseases. For example, microglia activated via NF-κβ increase superoxide production. The ROS generated in this process triggers cell death in neurons through lipid peroxidation and DNA damage. At the same time, this oxidative damage reactivates surrounding microglia, perpetuating the cycle. While ROS accumulation activates the NF-κB pathway, increasing inflammation, inflammatory cytokines such as TNF-α and IL-6 accelerate mitochondrial ROS production [19]. This interaction creates a positive feedback loop that triggers neuronal death. This bidirectional interaction acts as a “silent accelerator” in the progression of neurodegenerative diseases. Therefore, when developing treatment strategies, it is necessary to target inflammatory signaling pathways in addition to suppressing oxidative stress, requiring a dual-pronged approach.
Neuroinflammation and oxidative stress exhibit distinct yet partially convergent profiles across disease stages in ALS and AD. In ALS, the early stages are characterized by subtle yet detectable increases in oxidative stress markers within motor neurons, accompanied by microglial priming and mild astrocytic activation, often preceding overt motor symptoms. As ALS progresses, this evolves into a more pronounced proinflammatory milieu, with sustained microglial activation, elevated proinflammatory cytokines, and amplification of mitochondrial ROS production, creating a self-reinforcing OS–NI loop that accelerates motor neuron degeneration [56]. In AD, mild stages are typically associated with regionally restricted microglial activation around amyloid plaques and moderate oxidative damage affecting synapses and dendrites, correlating with early cognitive decline. In moderate-to-severe AD, neuroinflammation becomes more diffuse and sustained, oxidative damage extends to lipids, proteins, and nucleic acids, and the failure of endogenous antioxidant and resolution pathways further entrenches the chronic OS–NI cycle, paralleling extensive neuronal and synaptic loss [57].
8 Clinical and Therapeutic Perspective
The growing evidence that NI and OS play a central role in the pathogenesis of neurodegenerative diseases has led to a reassessment of currently FDA-approved agents, not only for their symptomatic effects but also for their neuroprotective potential [3]. Agents such as riluzole, edaravone, memantine, and acetylcholinesterase (AChE) inhibitors (AChEIs) (donepezil, rivastigmine, galantamine) interfere with key processes at different levels, including glutamate excitotoxicity, free-radical production, microglial activation, and proinflammatory cytokine release [58,59,60]. Research on the repurposing of FDA-approved Alzheimer’s drugs has shown that donepezil and memantine are the most effective at crossing the blood-brain barrier [61]. However, the generally modest clinical efficacy of these drugs highlights the limitations of single-target approaches and the need for multi-target strategies.
8.1 Riluzole: Excitotoxicity, Oxidative Stress, and Microglial Activation
Riluzole is a benzothiazole derivative that primarily reduces glutamatergic neurotransmission, blocks voltage-gated Na+ channels, and exerts a multimodal mechanism of action with neuroprotective/antioxidant properties [62,63]. Clinical data show that it provides a limited but meaningful extension of life span [64,65]. Beyond this, recent experimental studies have demonstrated that riluzole can not only reduce presynaptic glutamate release but also modulate OS and inflammatory markers [66,67]. However, several studies have reported that riluzole attenuates oxidative damage caused by ALS pathogenesis in neuronal cells [62]. Riluzole also has antioxidant properties in other disease models. Studies using riluzole-treated AD rat models have shown that riluzole exerts neuroprotective effects in AD by decreasing AChE activity and OS markers [68]. Riluzole treatment in ALS patients has been reported to increase survival and improve motor function [65]. In human or animal-derived neuronal and glial cell models, riluzole has been shown to reduce ROS production, strengthen antioxidant defenses, and suppress microglial activation and proinflammatory cytokine release [69]. These data suggest that riluzole’s modest effect observed in the clinic may be partially related to the partial disruption of the NI-OS cycle. Results from preclinical studies evaluating its impact on AD indicate that riluzole treatment reduces glutamate excitotoxicity, microglial and astrocyte activation, ROS, and OS, while increasing synaptic plasticity and improving learning and memory [70,71]. However, most of these mechanistic findings are at the preclinical level and need to be validated in prospective, biomarker-based clinical studies. Therefore, riluzole can be considered one of the “first-generation neuroprotective agents” that target both glutamate excitotoxicity and the inflammatory response from the current perspective.
8.2 Edaravone: Free Radical Scavenging Effect and Nrf2 Modulation
Edaravone is a molecule developed as a classic free-radical scavenger that directly neutralizes hydroxyl radicals and other ROS and is primarily approved for acute ischemic stroke. It has subsequently been introduced for use in ALS. Both clinical and experimental data show that edaravone reduces lipid peroxidation, limits oxidative damage to DNA and proteins, and thereby increases neuronal survival [72]. The literature indicates that edaravone’s effects are not limited to direct radical scavenging; it can also activate the Nrf2 pathway and NF-κB-mediated inflammation [73,74]. This dual effect positions edaravone as a “bridge” agent in the NI-OS-inflammation axis [75]. However, its effect on survival in ALS patients remains relatively limited, and response heterogeneity is notable. Due to its potential impact on OS and neurodegeneration, edaravone has been reported to reduce Aβ accumulation, tau phosphorylation, and mitochondrial dysfunction processes in preclinical studies on AD [76]. Edaravone treatment has been reported to reduce microglia activation, inhibit the NF-κB/MAPK pathway, and improve cognitive performance [77,78]. Although no completed clinical studies have evaluated edaravone treatment in AD patients, the Alzheimer Study Using Oral Edaravone (ASURE) aims to evaluate the safety of oral edaravone in AD patients, its effects on OS biomarkers, and its early cognitive/functional effects [79].
8.3 Memantine: NMDA Antagonism, Anti-Inflammatory Effects, and Potential New Indications
Memantine is a voltage- and usage-dependent N-methyl-D-aspartate (NMDA) receptor antagonist used in moderate-to-severe AD that limits excitotoxicity without completely suppressing physiological glutamatergic transmission [80,81]. Recent reviews suggest that memantine may have regulatory effects not only at the synaptic level but also on microglial activity and proinflammatory signaling pathways [80]. In experimental models, memantine has been reported to reduce microglial activation, TNF-α and IL-1β levels, and NF-κB activity in the NI response induced by lipopolysaccharide (LPS) or trauma; it also contributes to antioxidant defense by increasing SOD and total thiol levels [82,83]. The clinical data set is expanding with studies investigating the potential benefits of memantine in some neurological conditions other than AD (vascular dementia, traumatic brain injury, other neuroinflammatory conditions), but the level of evidence for these indications remains limited [84,85,86]. Memantine has been suggested to be effective in ALS because it can reduce glutamate toxicity in SOD1 (G93A) mice by antagonizing NMDA receptors [87,88]. The literature generally reports that patients tolerate memantine well. However, data from large-scale randomized controlled trials specifically targeting “dementia-related motor-cognitive processes” in these disease groups are not yet sufficient. This situation makes memantine a repurposable candidate for an anti-excitotoxic and anti-inflammatory agent.
8.4 Acetylcholinesterase Inhibitors: From Symptomatic Treatment to a Multitarget Approach
AChEIs, primarily donepezil, rivastigmine, and galantamine, have traditionally been used to improve symptoms in AD [89,90]. However, in recent years, there has been increasing evidence that these agents can suppress microglial activation via the cholinergic anti-inflammatory pathway, reduce OS markers, and partially preserve mitochondrial function [4,91]. Microglial activation has been linked to increased AChE, and AChEIs may suppress this process [91]. Another study specifically reported that donepezil treatment suppressed inflammasome (NLRP3) activation in microglial cells following LPS and Aβ stimulation [92]. It has been shown that galantamine and donepezil can promote neuronal survival through effects on nicotinic receptor modulation, sigma-1 receptor interactions, and antioxidant defense, independent of AChE inhibition [93,94,95]. It was traditionally accepted that AChEIs were drugs that only improved symptoms (memory, attention, daily life) by increasing acetylcholine, that they did not stop the disease, but only alleviated the symptoms. Studies show that AChEIs not only increase acetylcholine but also reduce microglial activation, suppress NF-κB and Nrf2 pathways, reduce OS, support mitochondrial function, and increase neurotrophic factors (especially galantamine) [91]. Therefore, AChEIs are now also considered multi-target drugs with neuroprotective potential that affect multiple pathological targets. These findings suggest that AChEIs can be reframed as components of multi-target neuroprotective strategies rather than “single-target symptomatic agents.” However, the disease-modifying effects of AChEIs at the clinical level remain unclear.
8.5 Combination Therapies and Multi-Targeted Approaches
The progression of NI and OS through intertwined networks limits the clinical efficacy of drugs developed based on the single-molecule–single-target logic. Consequently, several major trends have emerged over the past decade. Pharmacological combinations involving multiple drugs include the riluzole + edaravone combination in ALS treatment, which simultaneously targets excitotoxicity and OS [96]; the use of memantine with AChEIs [97]; Treatment and prevention strategies involving antioxidants or phytochemical agents (Origanum species, resveratrol, curcumin, etc.) [98,99] have shown synergistic effects at the preclinical level. However, randomized, controlled clinical trials remain insufficient.
The polypharmacology approach, in which a single drug affects multiple receptors or mechanisms via the development of multi-targeted, directed ligands (MTDLs), offers a promising treatment strategy that simultaneously modulates multiple interconnected pathways in AD pathology [100]. Polypharmacology is also trending in ALS treatment, as it is an effective way to address a multifactorial etiology involving excitotoxicity, mitochondrial dysfunction, OS, and microglial activation. Multi-target molecules, in which a single molecule is deliberately designed to affect multiple targets, particularly those derived from donepezil and memantine, aim to combine antioxidant, anti-inflammatory, and anti-amyloid properties within a single structure, in addition to AChE inhibition [101]. Recent reviews have reported that these hybrid molecules may offer a stronger neuroprotective profile by suppressing OS and microglial activity [102].
Current studies focus on small molecules, antibodies, and cell-based therapies aimed at reprogramming the microglial phenotype from an M1-like proinflammatory profile to an M2-like neuroprotective profile [16]. Targeting microglia and immunomodulatory approaches also aim to control microglia numbers through depletion and subsequent repopulation. Modulation of microglia is a new and promising area in AD and ALS. It is hoped that microglial modulation, when combined with strategies targeting OS and inflammation, may provide meaningful neuroprotection. Currently, there is no FDA-approved drug developed explicitly as a microglia modulator for neurodegenerative diseases. Existing FDA-approved medicines for neurodegenerative diseases were initially approved based on different mechanisms of action. Still, research indicates that some drugs may have secondary effects on microglia or are being actively investigated for this potential. The preclinical and clinical evidence, as well as the translational status, of pharmacological and immunomodulatory agents targeting microglial and neuroinflammatory pathways in AD and ALS are evaluated in Table 1.
Table 1: Pharmacological and immunomodulatory agents targeting microglial and neuroinflammatory pathways in Alzheimer’s disease and amyotrophic lateral sclerosis: clinical evidence and translational status.
| Therapy/Agent | Disease | Target Mechanism(s) | Pathway | Clinical Evidence Level | Clinical Efficacy Summary (Inline-Referenced) | Model/Population | Dose, Treatment Duration, Recovery Rate | Classification According to Clinical Maturity Level |
|---|---|---|---|---|---|---|---|---|
| Riluzole | ALS | Glutamate release inhibition | Excitotoxicity attenuation | Approved (ALS) | provides modest but reproducible survival and progression benefits in ALS and remains standard of care [64,103]. | ALS patients | 50 mg orally twice daily; chronic use | Approved/Clinically established treatments |
| Edaravone | ALS | Free-radical scavenging | Oxidative stress reduction | Moderate clinical evidence | Meta-analyses show statistically significant but clinically modest slowing of functional decline in selected ALS patients, while real-world studies do not support consistent long-term disease modification [13,104,105]. | ALS patients | 60 mg i.v; 6 cycles over 24 weeks | |
| Donepezil | AD (mild-severe) | AChE inhibition | Cholinergic signaling | Moderate-quality evidence | provides modest but statistically significant symptomatic benefits across AD stages, maintained only during active treatment, without disease modification [106,107]. | AD patients | 5–10 mg/day (23 mg ER explored); 12–50 weeks | |
| Memantine | AD (moderate–severe) | NMDA receptor antagonism | Glutamatergic signaling | High-certainty evidence | shows small symptomatic benefits in moderate-to-severe AD, but no benefit in mild AD or ALS, and no sustained post-treatment efficacy [108,109,110]. | AD patients; ALS patients | 20 mg/day; ~6–7 months (up to 40 weeks) optimal; recovery unclear | |
| Masitinib | AD (mild to moderate) | c-Kit and Lyn inhibition | NF-κB/NLRP3 modulation | Phase III | demonstrated modest cognitive improvement in mild-to-moderate AD in Phase III trials; disease-modifying effects remain unconfirmed [111]. | AD patients | 4.5 mg/kg/day; 24 weeks | Late phase/advanced clinical development |
| Usnoflast (ZYIL1) | ALS | NLRP3 inhibition | Inflammasome signaling | Phase II | Phase II data showed acceptable safety, with non-significant trends toward a slower decline in ALSFRS-R, supporting further study [112]. | ALS patients | 25–75 mg orally BID for 12 weeks | |
| Low-dose IL-2(Aldesleukin) | AD | Treg expansion | IL-2/Treg immunoregulation | Phase 2a | The Phase 2a study demonstrated safety, Treg expansion, and exploratory biomarker changes, but did not demonstrate definitive efficacy [113]. | AD patients | 106 IU/day SC in cycles; recovery not established | |
| Foralumab | Early AD | Intranasal anti-CD3 | T-cell immunomodulation | Phase II (ongoing) | Preclinical studies showed reduced neuroinflammation; a Phase II trial is ongoing, with no efficacy readout yet [114,115,116]. | Rodents; early AD patients | 50–100 μg intranasal cycles; efficacy pending | |
| AL002 | AD | TREM2 agonism | TREM2–DAP12 signaling | Preclinical, Phase I–II | Preclinical efficacy and Phase I target engagement were observed, but Phase II failed to meet the primary endpoint in early AD [117,118,119,120]. | Mice; humans | 15–60 mg/kg i.v; no clinical benefit demonstrated | Early phase/target validation studies |
| Baricitinib | AD | JAK1/JAK2 inhibition | JAK-STAT signaling | Exploratory trial | An exploratory AD trial demonstrated CSF penetration and biomarker modulation without a clinical efficacy assessment [121,122] | AD patients | 2–4 mg/day for 24 weeks; efficacy not established | |
| MCC950 | AD preclinical | NLRP3 inhibition | Inflammasome signaling | Preclinical | MCC950 reduced inflammasome activation and improved pathology in AD mouse models [123]. | Rodent models | ~10 mg/kg i.p.; preclinical only | Preclinical/translational candidates |
| Fingolimod | AD preclinical | S1P modulation | Immune trafficking reduction | Preclinical | Fingolimod reduced amyloid pathology and improved behavior in AD mouse models [124,125]. | AD mouse models | 0.03–1 mg/kg/day; preclinical | |
| Dimethyl fumarate (Tecfidera) | AD preclinical | NRF2 activation | NRF2/ARE pathway | Preclinical | Evidence for AD remains preclinical despite strong efficacy in MS [126,127]. | MS patients; AD models | 240 mg BID (MS); no AD dosing | |
| Omaveloxolone | AD preclinical | NRF2 activation | NRF2/ARE signaling | Preclinical | Omaveloxolone improved pathology and cognition in AD mouse models only [128]. | APP/PS1 mice | Preclinical regimens only | |
| Anakinra | NI | IL-1 receptor antagonism | IL-1 blockade | Phase II (ICH) | Clinical benefit shown in ICH but no evidence for AD efficacy [129]. | ICH patients | 100 mg/s.a short course | Mechanistic comparators (non-AD efficacy) |
| Rilonacept | IL-1 axis | IL-1 cytokine trap | IL-1 sequestration | Approved auto-inflammatory syndrome (non-AD) | No clinical evidence supports efficacy in AD [130]. | Humans | 320 mg loading then 160 mg weekly; no AD efficacy | |
| Canakinumab | IL-1β axis | Anti-IL-1β mAb | IL-1β blockade | Approved auto-inflammatory syndrome (non-AD) | No evidence supports efficacy in AD or other neurodegenerative diseases [131]. | Humans | 150 mg SC every 8 weeks; no AD efficacy | |
| Resveratrol | AD; ALS preclinical | SIRT1 activation | NF-κB modulation | Phase II: preclinical | Phase II AD trial showed safety and biomarker modulation without cognitive benefit; ALS efficacy limited to preclinical models [132,133,134]. | Humans; ALS models | 50–2000 mg/day (AD); preclinical ALS dosing | Nutraceutical/low translation success |
| Curcumin | AD; ALS | Anti-inflammatory | NF-κB inhibition | Phase II: preclinical | Clinical trials in AD and ALS failed to show a significant benefit despite promising preclinical data [135,136,137]. | Humans; animal models | 600 mg–4 g/day; 6–12 months; no recovery |
Candidates for microglia-targeted approaches include Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) targeting (AL002), IL-2 immunomodulation, masitinib (an oral tyrosine kinase inhibitor developed for cancer treatment), and CD3 modulation (foralumab) in AD (Table 1). TREM2 is a lipid receptor expressed on microglia and other tissue macrophages. It supports their survival and proliferation by transmitting activating signals into cells via the adaptor DAP12 [138]. When TREM2 is deficient, microglia cannot effectively cluster around Aβ, resulting in less compact plaques with longer, more branched amyloid fibrils. This increased plaque surface exposure directly correlates with more severe neuritic tau hyperphosphorylation and axonal dystrophy around amyloid deposits. The defective microglial response disrupts the formation of a neuroprotective barrier that normally regulates amyloid compaction and insulation, thereby exacerbating neuritic damage [139,140]. A study was planned to evaluate the efficacy of low-dose IL-2 in expanding regulatory T cells (Tregs) to modulate disease progression in AD patients, based on literature findings suggesting that Tregs may be a modifiable therapeutic target. Low-dose IL-2 treatment was reported to sustainably increase Treg numbers in AD biomarkers and clinical scales and improve Aβ42 levels. TREM2-associated microglia clustering around plaques is thought to be necessary to control Aβ plaques and associated neuropathology. In the study, no overall reduction in Aβ or increase in microglia clustering around Aβ plaques was observed in mice treated with AL002c; however, it was reported to reduce Aβ plaque toxicity and alleviate neuritic dystrophy associated with AD-related neuronal dysfunction [118]. The anti-CD3 antibody has been demonstrated to elicit regulatory immune mechanisms in the periphery and reduce microglial activation in neurological diseases, including AD [141,142]. Dimethyl fumarate (Tecfidera), an approved MS treatment, can affect glial cells and reduce inflammation in MMS. Dimethyl Fumarate has been shown to exert its effects by modulating NF-κB signaling through its Nrf2-activating properties and immunomodulatory activities [143,144]. Baricitinib, an FDA-approved JAK inhibitor for rheumatoid arthritis, is being evaluated in the literature for its potential effects on AD and ALS by inhibiting NI mediated through the Janus kinase/Signal transducer and activator of transcription 3 (JAK/STAT3) pathway. Treatment of AD rats with baricitinib has been shown to alleviate abnormal JAK2/STAT3 signaling pathway activation induced by ovariectomy/D-galactose, leading to significant reductions in p-JAK2 and p-STAT3 levels and activation of the AKT/PI3K/mTOR signaling, as well as reducing parameters of NI (e.g., NF-κB, TNF-α, and IL-6) and OS (e.g., iNOS and MDA) [145,146].
The activation of the NLRP3 inflammasome, which is essential for the maturation and release of proinflammatory cytokines, can lead to lysosomal damage through the accumulation of Aβ and Tau. In a study focusing on NLRP3-mediated NI, the underlying mechanisms of MCC950-mediated attenuation of AD-like pathology were investigated. The NLRP3 inflammasome is reported to initiate inflammatory responses by activating caspase-1 and promoting the release of proinflammatory cytokines such as IL-1β and IL-18. The study results indicate that MCC950 is an effective anti-inflammatory agent [111]. Masitinib is the strongest candidate for a microglia-targeted approach in ALS, but most strategies are currently at the preclinical level, and human clinical trials are limited. Masitinib is a tyrosine kinase inhibitor that targets mast cells and microglia cells. In a study in which masitinib was administered in addition to a cholinesterase inhibitor and/or memantine in patients with AD-related dementia, masitinib (4.5 mg/kg/day) was reported to improve overall cognitive function [147]. Fingolimod (Gilenya), a modulator of the sphingosine 1-phosphate receptor, is an FDA-approved drug for the treatment of multiple sclerosis. The literature indicates that fingolimod may suppress NI and convert microglia from a proinflammatory (M1) to an anti-inflammatory (M2) phenotype [148]. In a study in which fingolimod was administered intranasally to repolarize microglia and reduce Aβ expression, a ROS-responsive, carrier-free gene-delivery nanosystem (FTBR-NAC) was developed. Upon reaching the brain, FTBR responded to high ROS levels in the pathological region, enabling Fingolimod release. This application was reported to improve cognitive function in AD-mice [148]. Another study reported that, when administered orally to APP/PS1 mice for 2 months, fingolimod reduced NI, enhanced cognitive function, and alleviated brain Aβ accumulation. Furthermore, the treatment was reported to alleviate AD-related pathologies, including tau hyperphosphorylation, NI, and neurodegeneration [125]. Additionally, Fingolimod was shown to exert a protective effect via its immunomodulatory action in an experimental ALS model [149]. Safety has also been established in a phase-2 clinical trial in ALS patients [150]. Using publicly available transcriptomic data and bioinformatics analyses, a study of drug repurposing identified distinct gene expression profiles in SOD1G93A microglia and SOD1G93A motor neurons. Belinostat, auranofin, COT-10b, BRD-K78930611, and AZD-80552 were reported as candidates for ALS treatment, mimicking the protective state of microglia and reversing the toxic state of motor neurons. Researchers predict that establishing a link between the M2 state of microglia and drug repositioning could provide a new perspective on ALS pathophysiology [151].
Small-molecule therapies are promising; compounds such as metformin, rapamycin, and ketone bodies can modulate microglial metabolism [152]. Cell-based therapies involve pre-conditioned microglia and monocytes/macrophages that can migrate to damaged brain regions and release protective remodeling factors [153]. Evidence suggests these approaches are particularly important for stroke, AD, and Parkinson’s diseases, with researchers highlighting their potential to reduce neuroinflammation and enhance neuroprotection [154]. However, most studies are preliminary, and translational research is still needed to establish definitive clinical protocols. The heterogeneity of microglial cells, variation in microglial phenotype with disease stage, and differences between humans and mice are the main limitations that have prevented clinical translation so far. To advance in this area, it is essential to identify disease-specific strategies, use biomarkers to guide treatment, and understand microglial heterogeneity.
8.5.1 Microglial Phenotypic Transformation (M1/M2) and Drug Intervention Targets
Although microglial activation is traditionally defined by M1 and M2 phenotypes, it is increasingly recognized that the microglial response in neurodegenerative diseases is dynamic, disease-stage-dependent, and highly heterogeneous [155]. In the context of AD and ALS, key regulatory factors that determine microglial phenotypic transformation include innate immune sensors (e.g., the NLRP3 inflammasome) [156], NF-κB, phagocytosis, and disease-associated microglia (DAM) programs (TREM2-DAP12 signaling) [156,157,158], and redox balance [156]. Activation or suppression of these pathways is a critical factor determining whether microglial cells adopt a pro-inflammatory or more protective phenotype. Specifically, danger-related molecular signals, such as Aβ and tau accumulation, enhance the microglial inflammatory response, whereas TREM2-mediated signaling can support phagocytic capacity and cellular survival. Additionally, Nrf2 activation may exert an indirect regulatory effect on the microglia phenotype by weakening the vicious cycle between oxidative stress and inflammation [156,157]. These regulatory mechanisms offer specific therapeutic targets for microglia-targeted pharmacological interventions. TREM2 agonists aim to increase phagocytosis, NLRP3 inhibitors aim to suppress inflammasome activation, and agents targeting NF-κβ pathways aim to reduce pro-inflammatory cytokine production [156]. In addition, dimethyl fumarate, an immunomodulatory agent, is being evaluated as a potential therapeutic approach that may contribute to the indirect reprogramming of microglial activity and a more stable phenotypic profile [159,160].
8.5.2 Holistic Assessment and Translational Perspective
The findings discussed in this study reveal that microglial activation in neurodegenerative diseases cannot be explained solely by a binary M1/M2 phenotype distinction, but should be evaluated within a dynamic, continuous spectrum that depends on disease stage, tissue microenvironment, and metabolic status. In pathologies such as AD, ALS, and Parkinson’s disease, the microglia response is simultaneously shaped by numerous interconnected mechanisms, including innate immune sensors, inflammatory signaling, phagocytosis, and redox balance [161,162]. In this context, the NLRP3 inflammasome, NF-κB, and MAPK pathways stand out as the main regulators of the pro-inflammatory microglial response [51,163], while TREM2-DAP12 signaling supports phagocytosis capacity and cellular survival through DAM programs [164,165]. In addition, the Nrf2-mediated antioxidant response plays an indirect but critical role in the microglial phenotype as a high-level regulatory axis that limits the interaction between oxidative stress and inflammation [166,167]. Microglia-targeted therapeutic strategies aim to interfere with different nodes of this multi-layered regulatory network. NLRP3 inhibitors [168], small-molecule inhibitors targeting the JAK/STAT and NF-κB pathways [169], TREM2 agonists [170], and tyrosine kinase inhibitors [171] are among the prominent approaches for suppressing the inflammatory response and optimizing phagocytosis. However, pre-conditioned microglia or monocyte/macrophage-based cellular therapies with small molecules such as metformin and rapamycin targeting microglial metabolism offer a promising framework for phenotypic reprogramming [172,173]. However, much of the current data is based on preclinical studies, and the interspecies differences in human microglial biology pose significant limitations in translating these approaches into clinical practice. Therefore, future studies focusing on disease-stage-specific biomarkers, patient subgroup identification, and combinatorial treatment strategies are critical for the clinical success of microglia-targeted therapies.
8.6 Clinical Limitations and Future Trends
Although current FDA-approved agents demonstrate some regulatory effects on NI and OS, their disease-modifying effects are limited in most disease states and do not provide a radical treatment. The main reasons for this are the complex nature of neurodegenerative diseases, late diagnosis and consequently late treatment, the assumption of a ‘one-size-fits-all’ treatment approach for heterogeneous patient groups, the bioavailability of drugs, their affinity for the BBB, and the dynamic nature of the neuroinflammatory response (incomplete suppression can also be harmful). Therefore, the current trend is to focus on the following areas: New molecules that target the selective activation of Nrf2 while inhibiting NF-κB (e.g., omaveloxolone [129,174,175], dimethyl fumarate [176], diroximel fumarate [177]; canakinumab, rilonacept and anakinra, which are NLRP3 inflammasome inhibitors [178]; and microglia phenotype modulators; the personalised use of current agents based on biomarkers; and the rational combination of classical drugs with plant polyphenols and other antioxidants.
Inflammation and OS form a tightly interlinked pathological axis that drives neuronal vulnerability in ALS and AD. Mitochondrial dysfunction—particularly matrix-derived ROS production, mtDNA leakage, and activation of NLRP3 and cGAS–STING pathways—emerges as a central mechanism connecting these two processes. This review highlights the novelty of integrating these mitochondrial, redox, and innate immune pathways into a unified framework, offering a clearer understanding of how OS and NI co-evolve during neurodegeneration. Although current FDA-approved neuroprotective agents provide only partial modulation of these mechanisms, they reveal underrecognized effects on redox balance and inflammatory signaling that may guide rational drug repurposing. The most important insight of this work is the identification of shared molecular leverage points—such as mitochondrial ROS control, inflammasome inhibition, and restoration of antioxidant defenses—that could be targeted simultaneously to disrupt the OS–NI cycle. Together, these findings support the development of multi-targeted and combination therapeutic strategies as the most promising route toward achieving genuine disease-modifying effects in ALS and AD. In this context, based on the modeling methods used, a strategy can be developed to evaluate the efficacy and reliability of combination therapies with Memantine or Donepezil, which have strong CNS penetration, together with antioxidant/anti-inflammatory drugs. Future research should prioritize standardized experimental models, longitudinal analyses, and high-resolution mitochondrial and immunometabolic profiling to more precisely map the OS–NI cycle and identify actionable molecular checkpoints.
Acknowledgement:
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: Conceptualization, Aslı Aykaç; data acquisition, Aslı Aykaç, and Eda Becer; formal analysis, Aslı Aykaç, Eda Becer, and Ahmet Özer Şehirli; validation, Aslı Aykaç, Eda Becer, and Ahmet Özer Şehirli; data curation, Aslı Aykaç, Eda Becer, and Ahmet Özer Şehirli; supervision, Aslı Aykaç; writing—original draft, Aslı Aykaç; writing—review & editing, Aslı Aykaç, Eda Becer, and Ahmet Özer Şehirli. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: No new data were created or analyzed in this study. Data sharing is not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest.
References
1. Kovacs GG . Molecular pathology of neurodegenerative diseases: Principles and practice. J Clin Pathol. 2019; 72( 11): 725– 35. doi:10.1136/jclinpath-2019-205952. [Google Scholar] [CrossRef]
2. Gadhave DG , Sugandhi VV , Jha SK , Nangare SN , Gupta G , Singh SK , et al. Neurodegenerative disorders: Mechanisms of degeneration and therapeutic approaches with their clinical relevance. Ageing Res Rev. 2024; 99: 102357. doi:10.1016/j.arr.2024.102357. [Google Scholar] [CrossRef]
3. Zhang W , Xiao D , Mao Q , Xia H . Role of neuroinflammation in neurodegeneration development. Signal Transduct Target Ther. 2023; 8( 1): 267. doi:10.1038/s41392-023-01486-5. [Google Scholar] [CrossRef]
4. Dash UC , Bhol NK , Swain SK , Samal RR , Nayak PK , Raina V , et al. Oxidative stress and inflammation in the pathogenesis of neurological disorders: Mechanisms and implications. Acta Pharm Sin B. 2025; 15( 1): 15– 34. doi:10.1016/j.apsb.2024.10.004. [Google Scholar] [CrossRef]
5. Zhang K , Wen M , Nan X , Zhao S , Li H , Ai Y , et al. NMDA receptors in neurodegenerative diseases: Mechanisms and emerging therapeutic strategies. Front Aging Neurosci. 2025; 17: 1604378. doi:10.3389/fnagi.2025.1604378. [Google Scholar] [CrossRef]
6. Teleanu DM , Niculescu AG , Lungu II , Radu CI , Vladâcenco O , Roza E , et al. An overview of oxidative stress, neuroinflammation, and neurodegenerative diseases. Int J Mol Sci. 2022; 23( 11): 5938. doi:10.3390/ijms23115938. [Google Scholar] [CrossRef]
7. Cha SJ , Kim K . Effects of the edaravone, a drug approved for the treatment of amyotrophic lateral sclerosis, on mitochondrial function and neuroprotection. Antioxidants. 2022; 11( 2): 195. doi:10.3390/antiox11020195. [Google Scholar] [CrossRef]
8. Jiang J , Wang Y , Deng M . New developments and opportunities in drugs being trialed for amyotrophic lateral sclerosis from 2020 to 2022. Front Pharmacol. 2022; 13: 1054006. doi:10.3389/fphar.2022.1054006. [Google Scholar] [CrossRef]
9. Al-Horani RA . Riluzole and its prodrugs for the treatment of Alzheimer’s disease. Pharm Pat Anal. 2023; 12( 2): 79– 85. doi:10.4155/ppa-2023-0001. [Google Scholar] [CrossRef]
10. Berríos-Cárcamo P , Quezada M , Quintanilla ME , Morales P , Ezquer M , Herrera-Marschitz M , et al. Oxidative stress and neuroinflammation as a pivot in drug abuse. A focus on the therapeutic potential of antioxidant and anti-inflammatory agents and biomolecules. Antioxidants. 2020; 9( 9): 830. doi:10.3390/antiox9090830. [Google Scholar] [CrossRef]
11. Wohleb ES , Godbout JP . Basic aspects of the immunology of neuroinflammation. Mod Trends Pharmacopsychiatry. 2013; 28: 1– 19. doi:10.1159/000343964. [Google Scholar] [CrossRef]
12. Colombo E , Farina C . Astrocytes: Key regulators of neuroinflammation. Trends Immunol. 2016; 37( 9): 608– 20. doi:10.1016/j.it.2016.06.006. [Google Scholar] [CrossRef]
13. Kraft AD , Harry GJ . Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. Int J Environ Res Public Health. 2011; 8( 7): 2980– 3018. doi:10.3390/ijerph8072980. [Google Scholar] [CrossRef]
14. Block ML , Zecca L , Hong JS . Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat Rev Neurosci. 2007; 8( 1): 57– 69. doi:10.1038/nrn2038. [Google Scholar] [CrossRef]
15. Colonna M , Butovsky O . Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017; 35: 441– 68. doi:10.1146/annurev-immunol-051116-052358. [Google Scholar] [CrossRef]
16. Gao C , Jiang J , Tan Y , Chen S . Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct Target Ther. 2023; 8( 1): 359. doi:10.1038/s41392-023-01588-0. [Google Scholar] [CrossRef]
17. Bivona G , Iemmolo M , Agnello L , Lo Sasso B , Gambino CM , Giglio RV , et al. Microglial activation and priming in Alzheimer’s disease: State of the art and future perspectives. Int J Mol Sci. 2023; 24( 1): 884. doi:10.3390/ijms24010884. [Google Scholar] [CrossRef]
18. Miao J , Ma H , Yang Y , Liao Y , Lin C , Zheng J , et al. Microglia in Alzheimer’s disease: Pathogenesis, mechanisms, and therapeutic potentials. Front Aging Neurosci. 2023; 15: 1201982. doi:10.3389/fnagi.2023.1201982. [Google Scholar] [CrossRef]
19. Simpson DSA , Oliver PL . ROS generation in microglia: Understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants. 2020; 9( 8): 743. doi:10.3390/antiox9080743. [Google Scholar] [CrossRef]
20. Heneka MT , van der Flier WM , Jessen F , Hoozemanns J , Thal DR , Boche D , et al. Neuroinflammation in Alzheimer disease. Nat Rev Immunol. 2025; 25( 5): 321– 52. doi:10.1038/s41577-024-01104-7. [Google Scholar] [CrossRef]
21. Iliyasu MO , Musa SA , Oladele SB , Iliya AI . Amyloid-beta aggregation implicates multiple pathways in Alzheimer’s disease: Understanding the mechanisms. Front Neurosci. 2023; 17: 1081938. doi:10.3389/fnins.2023.1081938. [Google Scholar] [CrossRef]
22. Kim GH , Kim JE , Rhie SJ , Yoon S . The role of oxidative stress in neurodegenerative diseases. Exp Neurobiol. 2015; 24( 4): 325– 40. doi:10.5607/en.2015.24.4.325. [Google Scholar] [CrossRef]
23. Misrani A , Tabassum S , Yang L . Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease. Front Aging Neurosci. 2021; 13: 617588. doi:10.3389/fnagi.2021.617588. [Google Scholar] [CrossRef]
24. Jomova K , Raptova R , Alomar SY , Alwasel SH , Nepovimova E , Kuca K , et al. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch Toxicol. 2023; 97( 10): 2499– 574. doi:10.1007/s00204-023-03562-9. [Google Scholar] [CrossRef]
25. Peggion C , Calì T , Brini M . Mitochondria dysfunction and neuroinflammation in neurodegeneration: Who comes first? Antioxidants. 2024; 13( 2): 240. doi:10.3390/antiox13020240. [Google Scholar] [CrossRef]
26. Ma Y , Song R , Duan C . Mitochondrial quality control and transfer communication in neurological disorders and neuroinflammation. Front Immunol. 2025; 16: 1542369. doi:10.3389/fimmu.2025.1542369. [Google Scholar] [CrossRef]
27. Lingappan K . NF-κB in oxidative stress. Curr Opin Toxicol. 2018; 7: 81– 6. doi:10.1016/j.cotox.2017.11.002. [Google Scholar] [CrossRef]
28. Villavicencio Tejo F , Quintanilla RA . Contribution of the Nrf2 pathway on oxidative damage and mitochondrial failure in parkinson and Alzheimer’s disease. Antioxidants. 2021; 10( 7): 1069. doi:10.3390/antiox10071069. [Google Scholar] [CrossRef]
29. Riley JS , Tait SW . Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020; 21: e49799. doi:10.15252/embr.201949799. [Google Scholar] [CrossRef]
30. Marchi S , Guilbaud E , Tait SWG , Yamazaki T , Galluzzi L . Mitochondrial control of inflammation. Nat Rev Immunol. 2023; 23( 3): 159– 73. doi:10.1038/s41577-022-00760-x. [Google Scholar] [CrossRef]
31. Tschopp J , Schroder K . NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010; 10( 3): 210– 5. doi:10.1038/nri2725. [Google Scholar] [CrossRef]
32. Liu J , Wang S , Fan L , Zhou X , Zhang S , Wang Q , et al. Protein regulatory network mediated by palmitoylation modifications in the pathological progression of Parkinson’s disease: A narrative review. Front Immunol. 2025; 16: 1615001. doi:10.3389/fimmu.2025.1615001. [Google Scholar] [CrossRef]
33. Tian Y , Merkwirth C , Dillin A . Mitochondrial UPR: A double-edged sword. Trends Cell Biol. 2016; 26( 8): 563– 5. doi:10.1016/j.tcb.2016.06.006. [Google Scholar] [CrossRef]
34. Gong Z , Pan J , Shen Q , Li M , Peng Y . Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J Neuroinflamm. 2018; 15( 1): 242. doi:10.1186/s12974-018-1282-6. [Google Scholar] [CrossRef]
35. Xu X , Pang Y , Fan X . Mitochondria in oxidative stress, inflammation and aging: From mechanisms to therapeutic advances. Signal Transduct Target Ther. 2025; 10( 1): 190. doi:10.1038/s41392-025-02253-4. [Google Scholar] [CrossRef]
36. Fang EF , Hou Y , Palikaras K , Adriaanse BA , Kerr JS , Yang B , et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019; 22( 3): 401– 12. doi:10.1038/s41593-018-0332-9. [Google Scholar] [CrossRef]
37. Morgan MJ , Liu ZG . Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011; 21( 1): 103– 15. doi:10.1038/cr.2010.178. [Google Scholar] [CrossRef]
38. Ahmed SM , Luo L , Namani A , Wang XJ , Tang X . Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim Biophys Acta Mol Basis Dis. 2017; 1863( 2): 585– 97. doi:10.1016/j.bbadis.2016.11.005. [Google Scholar] [CrossRef]
39. Barbalho SM , Leme Boaro B , da Silva Camarinha Oliveira J , Patočka J , Barbalho Lamas C , Tanaka M , et al. Molecular mechanisms underlying neuroinflammation ıntervention with medicinal plants: A critical and narrative review of the current literature. Pharmaceuticals. 2025; 18( 1): 133. doi:10.3390/ph18010133. [Google Scholar] [CrossRef]
40. Oeckinghaus A , Ghosh S . The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009; 1( 4): a000034. doi:10.1101/cshperspect.a000034. [Google Scholar] [CrossRef]
41. Varsamos I , Patilas C , Galanis A , Zachariou D , Tsalimas G , Sakellariou E , et al. The Impact of Nuclear Factor Kappa B on the Response of Microglia in Spinal Cord Injuries. Cureus. 2025; 17( 2): e79367. doi:10.7759/cureus.79367. [Google Scholar] [CrossRef]
42. Jong Huat T , Camats-Perna J , Newcombe EA , Onraet T , Campbell D , Sucic JT , et al. The impact of astrocytic NF-κB on healthy and Alzheimer’s disease brains. Sci Rep. 2024; 14( 1): 14305. doi:10.1038/s41598-024-65248-1. [Google Scholar] [CrossRef]
43. Cunningham A , Barrett E , Risch S , Lee PHU , Lee C , Moghekar A , et al. NFκB1: A common biomarker linking Alzheimer’s and Parkinson’s disease pathology. Front Neurosci. 2025; 19: 1589857. doi:10.3389/fnins.2025.1589857. [Google Scholar] [CrossRef]
44. Ngo V , Duennwald ML . Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants. 2022; 11( 12): 2345. doi:10.3390/antiox11122345. [Google Scholar] [CrossRef]
45. George M , Tharakan M , Culberson J , Reddy AP , Reddy PH . Role of Nrf2 in aging, Alzheimer’s and other neurodegenerative diseases. Ageing Res Rev. 2022; 82: 101756. doi:10.1016/j.arr.2022.101756. [Google Scholar] [CrossRef]
46. Obata T , Brown GE , Yaffe MB . MAP kinase pathways activated by stress: The p38 MAPK pathway. Crit Care Med. 2000; 28( 4 Suppl): N67– 77. doi:10.1097/00003246-200004001-00008. [Google Scholar] [CrossRef]
47. Kim EK , Choi EJ . Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010; 1802( 4): 396– 405. doi:10.1016/j.bbadis.2009.12.009. [Google Scholar] [CrossRef]
48. Forrester SJ , Kikuchi DS , Hernandes MS , Xu Q , Griendling KK . Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018; 122( 6): 877– 902. doi:10.1161/CIRCRESAHA.117.311401. [Google Scholar] [CrossRef]
49. Veglianese P , Lo Coco D , Bao Cutrona M , Magnoni R , Pennacchini D , Pozzi B , et al. Activation of the p38MAPK cascade is associated with upregulation of TNF alpha receptors in the spinal motor neurons of mouse models of familial ALS. Mol Cell Neurosci. 2006; 31( 2): 218– 31. doi:10.1016/j.mcn.2005.09.009. [Google Scholar] [CrossRef]
50. Jha SK , Jha NK , Kar R , Ambasta RK , Kumar P . p38 MAPK and PI3K/AKT signalling cascades in Parkinson’s disease. Int J Mol Cell Med. 2015; 4( 2): 67– 86. [Google Scholar]
51. Kelley N , Jeltema D , Duan Y , He Y . The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int J Mol Sci. 2019; 20( 13): 3328. doi:10.3390/ijms20133328. [Google Scholar] [CrossRef]
52. Holbrook JA , Jarosz-Griffiths HH , Caseley E , Lara-Reyna S , Poulter JA , Williams-Gray CH , et al. Neurodegenerative disease and the NLRP3 inflammasome. Front Pharmacol. 2021; 12: 643254. doi:10.3389/fphar.2021.643254. [Google Scholar] [CrossRef]
53. Blyufer A , Lhamo S , Tam C , Tariq I , Thavornwatanayong T , Mahajan SS . Riluzole: A neuroprotective drug with potential as a novel anti-cancer agent (Review). Int J Oncol. 2021; 59( 5): 95. doi:10.3892/ijo.2021.5275. [Google Scholar] [CrossRef]
54. Beal MF , Ferrante RJ , Browne SE , Matthews RT , Kowall NW , Brown RH Jr . Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol. 1997; 42( 4): 644– 54. doi:10.1002/ana.410420416. [Google Scholar] [CrossRef]
55. Schieber M , Chandel NS . ROS function in redox signaling and oxidative stress. Curr Biol. 2014; 24( 10): R453– 62. doi:10.1016/j.cub.2014.03.034. [Google Scholar] [CrossRef]
56. Stacchiotti C , Mazzella di Regnella S , Cinotti M , Spalloni A , Volpe E . Neuroinflammation and amyotrophic lateral sclerosis: Recent advances in anti-ınflammatory cytokines as therapeutic strategies. Int J Mol Sci. 2025; 26( 8): 3854. doi:10.3390/ijms26083854. [Google Scholar] [CrossRef]
57. Weinstock M . Role of oxidative stress and neuroinflammation in the etiology of Alzheimer’s disease: Therapeutic options. Antioxidants. 2025; 14( 7): 769. doi:10.3390/antiox14070769. [Google Scholar] [CrossRef]
58. Cheah BC , Vucic S , Krishnan AV , Kiernan MC . Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr Med Chem. 2010; 17( 18): 1942– 199. doi:10.2174/092986710791163939. [Google Scholar] [CrossRef]
59. Li SM , Mo MS , Xu PY . Progress in mechanisms of acetylcholinesterase inhibitors and memantine for treatment of Alzheimer’s disease. Neurosciences. 2015; 2: 274– 80. doi:10.4103/2347-8659.167305. [Google Scholar] [CrossRef]
60. Takatori Y . Mechanisms of neuroprotective effects of therapeutic acetylcholinesterase inhibitors used in treatment of Alzheimer’s disease. Yakugaku Zasshi. 2006; 126( 8): 607– 16. (In Japanese). doi:10.1248/yakushi.126.607. [Google Scholar] [CrossRef]
61. Akgüller Ö , Balcı MA , Cioca G . Network-medicine-guided drug repurposing for Alzheimer’s disease: A multi-dimensional systems pharmacology approach. Int J Mol Sci. 2025; 26( 20): 10003. doi:10.3390/ijms262010003. [Google Scholar] [CrossRef]
62. Bellingham MC . A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: What have we learned in the last decade? CNS Neurosci Ther. 2011; 17( 1): 4– 31. doi:10.1111/j.1755-5949.2009.00116.x. [Google Scholar] [CrossRef]
63. Sala G , Arosio A , Conti E , Beretta S , Lunetta C , Riva N , et al. Riluzole selective antioxidant effects in cell models expressing amyotrophic lateral sclerosis endophenotypes. Clin Psychopharmacol Neurosci. 2019; 17( 3): 438– 42. doi:10.9758/cpn.2019.17.3.438. [Google Scholar] [CrossRef]
64. Hinchcliffe M , Smith A . Riluzole: Real-world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degener Neurol Neuromuscul Dis. 2017; 7: 61– 70. doi:10.2147/DNND.S135748. [Google Scholar] [CrossRef]
65. Fang T , Al Khleifat A , Meurgey JH , Jones A , Leigh PN , Bensimon G , et al. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: A retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018; 17( 5): 416– 22. doi:10.1016/S1474-4422(18)30054-1. [Google Scholar] [CrossRef]
66. Vallée A , Vallée JN , Guillevin R , Lecarpentier Y . Riluzole: A therapeutic strategy in Alzheimer’s disease by targeting the WNT/β-catenin pathway. Aging. 2020; 12( 3): 3095– 113. doi:10.18632/aging.102830. [Google Scholar] [CrossRef]
67. Rotolo RA , Demuro J , Drummond G , Little C , Johns LD , Betz AJ . Prophylactic exposure to oral riluzole reduces the clinical severity and immune-related biomarkers of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2021; 356: 577603. doi:10.1016/j.jneuroim.2021.577603. [Google Scholar] [CrossRef]
68. Mokhtari Z , Baluchnejadmojarad T , Nikbakht F , Mansouri M , Roghani M . Riluzole ameliorates learning and memory deficits in Aβ25-35-induced rat model of Alzheimer’s disease and is independent of cholinoceptor activation. Biomed Pharmacother. 2017; 87: 135– 44. doi:10.1016/j.biopha.2016.12.067. [Google Scholar] [CrossRef]
69. Hendricus Maes KJ , Briedé JJ . Repurposing immunomodulatory drugs targeting microglia for amyotrophic lateral sclerosis. Brain Res. 2025; 1870: 150032. doi:10.1016/j.brainres.2025.150032. [Google Scholar] [CrossRef]
70. Lesuis SL , Kaplick PM , Lucassen PJ , Krugers HJ . Treatment with the glutamate modulator riluzole prevents early life stress-induced cognitive deficits and impairments in synaptic plasticity in APPswe/PS1dE9 mice. Neuropharmacology. 2019; 150: 175– 83. doi:10.1016/j.neuropharm.2019.02.023. [Google Scholar] [CrossRef]
71. Okamoto M , Gray JD , Larson CS , Kazim SF , Soya H , McEwen BS , et al. Riluzole reduces amyloid beta pathology, improves memory, and restores gene expression changes in a transgenic mouse model of early-onset Alzheimer’s disease. Transl Psychiatry. 2018; 8( 1): 153. Erratum in: Transl Psychiatry. 2019;9(1):61. doi:10.1038/s41398-018-0201-z. [Google Scholar] [CrossRef]
72. Dakroub F , Awada B , Abdelhady S , Shaito AA , Eid AH , Walker J , et al. Edaravone: Advances on cytoprotective effects, pharmacological properties, and mechanisms of action. Pharmacol Rev. 2025; 78( 1): 100101. doi:10.1016/j.pharmr.2025.100101. [Google Scholar] [CrossRef]
73. Edaravone Acute Infarction Study Group . Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction. Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc Dis. 2003; 15( 3): 222– 9. doi:10.1159/000069318. [Google Scholar] [CrossRef]
74. Zhang H , Zhu C , Zhou X , Wang L , Deng L , He B , et al. Edaravone dexborneol protected neurological function by targeting NRF2/ARE and NF-κB/AIM2 pathways in cerebral ischemia/reperfusion injury. Front Pharmacol. 2025; 16: 1581320. doi:10.3389/fphar.2025.1581320. [Google Scholar] [CrossRef]
75. Yoshino H . Edaravone for the treatment of amyotrophic lateral sclerosis. Expert Rev Neurother. 2019; 19( 3): 185– 93. doi:10.1080/14737175.2019.1581610. [Google Scholar] [CrossRef]
76. Jiao SS , Yao XQ , Liu YH , Wang QH , Zeng F , Lu JJ , et al. Edaravone alleviates Alzheimer’s disease-type pathologies and cognitive deficits. Proc Natl Acad Sci U S A. 2015; 112( 16): 5225– 30. doi:10.1073/pnas.1422998112. [Google Scholar] [CrossRef]
77. Xu C , Mei Y , Yang R , Luo Q , Zhang J , Kou X , et al. Edaravone Dexborneol mitigates pathology in animal and cell culture models of Alzheimer’s disease by inhibiting neuroinflammation and neuronal necroptosis. Cell Biosci. 2024; 14( 1): 55. doi:10.1186/s13578-024-01230-8. [Google Scholar] [CrossRef]
78. Wang HM , Zhang T , Huang JK , Xiang JY , Chen JJ , Fu JL , et al. Edaravone Attenuates the Proinflammatory Response in Amyloid-β-Treated Microglia by Inhibiting NLRP3 Inflammasome-Mediated IL-1β Secretion. Cell Physiol Biochem. 2017; 43( 3): 1113– 25. doi:10.1159/000481753. [Google Scholar] [CrossRef]
79. Oosthoek M , Lili A , Almeida A , van Loosbroek O , van der Geest R , de Greef-van der Sandt I , et al. ASURE clinical trial protocol: A randomized, placebo-controlled, proof-of-concept study aiming to evaluate safety and target engagement following administration of TW001 in early Alzheimer’s disease patients. J Prev Alzheimers Dis. 2023; 10( 4): 669– 74. doi:10.14283/jpad.2023.107. [Google Scholar] [CrossRef]
80. Lau CG , Zukin RS . NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007; 8: 413– 26. doi:10.1038/nrn2153. [Google Scholar] [CrossRef]
81. Kutzing MK , Luo V , Firestein BL . Protection from glutamate-induced excitotoxicity by memantine. Ann Biomed Eng. 2012; 40( 5): 1170– 81. doi:10.1007/s10439-011-0494-z. [Google Scholar] [CrossRef]
82. Choi H , Hwang S , Cho H , Ahn S , Yun HY , Song JS . Memantine modulates neuroinflammation and motor coordination in a Parkinson’s disease model. Brain Res. 2025; 1869: 150034. doi:10.1016/j.brainres.2025.150034. [Google Scholar] [CrossRef]
83. Bardaghi Z , Rajabian A , Beheshti F , Arabi MH , Hosseini M , Salmani H . Memantine, an NMDA receptor antagonist, protected the brain against the long-term consequences of sepsis in mice. Life Sci. 2023; 323: 121695. doi:10.1016/j.lfs.2023.121695. [Google Scholar] [CrossRef]
84. Wang Z , He X , Fan X . Postnatal administration of memantine rescues TNF-α-induced decreased hippocampal precursor proliferation. Neurosci Lett. 2018; 662: 173– 80. doi:10.1016/j.neulet.2017.10.022. [Google Scholar] [CrossRef]
85. Wu HM , Tzeng NS , Qian L , Wei SJ , Hu X , Chen SH , et al. Novel neuroprotective mechanisms of memantine: Increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology. 2009; 34( 10): 2344– 57. doi:10.1038/npp.2009.64. [Google Scholar] [CrossRef]
86. Wang F , Zou Z , Gong Y , Yuan D , Chen X , Sun T . Regulation of human brain microvascular endothelial cell adhesion and barrier functions by memantine. J Mol Neurosci. 2017; 62( 1): 123– 9. doi:10.1007/s12031-017-0917-x. [Google Scholar] [CrossRef]
87. Wang R , Zhang D . Memantine prolongs survival in an amyotrophic lateral sclerosis mouse model. Eur J Neurosci. 2005; 22( 9): 2376– 80. doi:10.1111/j.1460-9568.2005.04431.x. [Google Scholar] [CrossRef]
88. Tortarolo M , Grignaschi G , Calvaresi N , Zennaro E , Spaltro G , Colovic M , et al. Glutamate AMPA receptors change in motor neurons of SOD1G93A transgenic mice and their inhibition by a noncompetitive antagonist ameliorates the progression of amytrophic lateral sclerosis-like disease. J Neurosci Res. 2006; 83( 1): 134– 46. doi:10.1002/jnr.20715. [Google Scholar] [CrossRef]
89. Marucci G , Buccioni M , Ben DD , Lambertucci C , Volpini R , Amenta F . Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology. 2021; 190: 108352. doi:10.1016/j.neuropharm.2020.108352. [Google Scholar] [CrossRef]
90. Aykaç A , Özbeyli D , Pekol G , Şehirli AÖ . Evaluation of the effects of donepezil, memantine and α-lipoic acid combined administration in amnesia rats on impaired cognitive functions in terms of behavioural, apoptotic, cholinergic and glutamatergic systems. Clin Exp Health Sci. 2021; 11( 4): 733– 41. doi:10.33808/clinexphealthsci.856459. [Google Scholar] [CrossRef]
91. Walczak-Nowicka ŁJ , Herbet M . Acetylcholinesterase Inhibitors in the treatment of neurodegenerative diseases and the role of acetylcholinesterase in their pathogenesis. Int J Mol Sci. 2021; 22( 17): 9290. doi:10.3390/ijms22179290. [Google Scholar] [CrossRef]
92. Kim J , Lee HJ , Park SK , Park JH , Jeong HR , Lee S , et al. Donepezil Regulates LPS and Aβ-stimulated neuroinflammation through MAPK/NLRP3 inflammasome/STAT3 signaling. Int J Mol Sci. 2021; 22( 19): 10637. doi:10.3390/ijms221910637. [Google Scholar] [CrossRef]
93. Kume T , Sugimoto M , Takada Y , Yamaguchi T , Yonezawa A , Katsuki H , et al. Up-regulation of nicotinic acetylcholine receptors by central-type acetylcholinesterase inhibitors in rat cortical neurons. Eur J Pharmacol. 2005; 527( 1–3): 77– 85. doi:10.1016/j.ejphar.2005.10.028. [Google Scholar] [CrossRef]
94. Tsvetkova D , Obreshkova D , Zheleva-Dimitrova D , Saso L . Antioxidant activity of galantamine and some of its derivatives. Curr Med Chem. 2013; 20( 36): 4595– 608. doi:10.2174/09298673113209990148. [Google Scholar] [CrossRef]
95. Koola MM , Praharaj SK , Pillai A . Galantamine-memantine combination as an antioxidant treatment for schizophrenia. Curr Behav Neurosci Rep. 2019; 6( 2): 37– 50. doi:10.1007/s40473-019-00174-5. [Google Scholar] [CrossRef]
96. Jaiswal MK . Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med Res Rev. 2019; 39( 2): 733– 48. doi:10.1002/med.21528. [Google Scholar] [CrossRef]
97. Glinz D , Gloy VL , Monsch AU , Kressig RW , Patel C , McCord KA , et al. Acetylcholinesterase inhibitors combined with memantine for moderate to severe Alzheimer’s disease: A meta-analysis. Swiss Med Wkly. 2019; 149: w20093. doi:10.4414/smw.2019.20093. [Google Scholar] [CrossRef]
98. Libro R , Giacoppo S , Soundara Rajan T , Bramanti P , Mazzon E . Natural phytochemicals in the treatment and prevention of dementia: An overview. Molecules. 2016; 21( 4): 518. doi:10.3390/molecules21040518. [Google Scholar] [CrossRef]
99. Ansari F , Sohel M , Haidary MMH , Mostaq MS , Akter S , Nahar A , et al. Therapeutic potential of clinically proven natural products in the management of dementia. Heliyon. 2024; 10( 6): e27233. doi:10.1016/j.heliyon.2024.e27233. [Google Scholar] [CrossRef]
100. Kumar S , Ganesh P , Patil P , Ganesan RR , Urolagin D , Raghavendra NM . Polypharmacology in Alzheimer’s disease: Integrating AI, network pharmacology, and experimental validation. Int J Environ Sci. 2025; 359( 11): 24. doi:10.64252/v0k8cn36. [Google Scholar] [CrossRef]
101. Piemontese L , Tomás D , Hiremathad A , Capriati V , Candeias E , Cardoso SM , et al. Donepezil structure-based hybrids as potential multifunctional anti-Alzheimer’s drug candidates. J Enzyme Inhib Med Chem. 2018; 33( 1): 1212– 24. doi:10.1080/14756366.2018.1491564. [Google Scholar] [CrossRef]
102. Angelova VT , Stoyanov BP , Simeonova R . New Insights into the development of donepezil-based hybrid and natural molecules as multi-target drug agents for Alzheimer’s disease treatment. Molecules. 2024; 29( 22): 5314. doi:10.3390/molecules29225314. [Google Scholar] [CrossRef]
103. Bensimon G , Lacomblez L , Meininger V . A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994; 330( 9): 585– 91. doi:10.1056/NEJM199403033300901. [Google Scholar] [CrossRef]
104. Nourelden AZ , Kamal I , Hagrass AI , Tawfik AG , Elhady MM , Fathallah AH , et al. Safety and efficacy of edaravone in patients with amyotrophic lateral sclerosis: A systematic review and meta-analysis. Neurol Sci. 2023; 44( 10): 3429– 42. doi:10.1007/s10072-023-06869-8. [Google Scholar] [CrossRef]
105. Witzel S , Maier A , Steinbach R , Grosskreutz J , Koch JC , Sarikidi A , et al. Safety and effectiveness of long-term ıntravenous administration of edaravone for treatment of patients with amyotrophic lateral sclerosis. JAMA Neurol. 2022; 79( 2): 121– 30. Erratum in: JAMA Neurol. 2022;79(7):727 doi:10.1001/jamaneurol.2021.4893. [Google Scholar] [CrossRef]
106. Birks JS , Harvey RJ . Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev. 2018; 6( 6): CD001190. doi:10.1002/14651858.CD001190.pub3. [Google Scholar] [CrossRef]
107. Jelic V , Darreh-Shori T . Donepezil: A review of pharmacological characteristics and role in the management of Alzheimer disease. Clinical Medicine Insights: Therapeutics. 2010; 2: 771– 88. doi:10.4137/CMT.S5410. [Google Scholar] [CrossRef]
108. McShane R , Westby MJ , Roberts E , Minakaran N , Schneider L , Farrimond LE , et al. Memantine for dementia. Cochrane Database Syst Rev. 2019; 3( 3): CD003154. doi:10.1002/14651858.CD003154.pub6. [Google Scholar] [CrossRef]
109. de Carvalho M , Pinto S , Costa J , Evangelista T , Ohana B , Pinto A . A randomized, placebo-controlled trial of memantine for functional disability in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010; 11( 5): 456– 60. doi:10.3109/17482968.2010.498521. [Google Scholar] [CrossRef]
110. Bhai S , Levine T , Moore D , Bowser R , Heim AJ , Walsh M , et al. A 40-week phase 2B randomized, multicenter, double-blind, placebo-controlled study evaluating the safety and efficacy of memantine in amyotrophic lateral sclerosis. Muscle Nerve. 2025; 71( 1): 63– 72. doi:10.1002/mus.28287. [Google Scholar] [CrossRef]
111. Dubois B , López-Arrieta J , Lipschitz S , Doskas T , Spiru L , Moroz S , et al. Masitinib for mild-to-moderate Alzheimer’s disease: Results from a randomized, placebo-controlled, phase 3, clinical trial. Alzheimers Res Ther. 2023; 15( 1): 39. Erratum in: Alzheimers Res Ther. 2023;15(1):85. doi: 10.1186/s13195-023-01169-x. [Google Scholar] [CrossRef]
112. Yeole A , Khanna L , Doshi M , Sharma A , Uttarwar P , Doshi S , et al. A phase 2, proof-of-concept, placebo-controlled, randomized, multicenter, double-blind study to evaluate the efficacy, safety, tolerability, pharmacokinetics, and pharmacodynamics of Usnoflast (ZYIL1) in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Front Degener. 2025; 26( 7–8): 794– 801. doi:10.1080/21678421.2025.2515900. [Google Scholar] [CrossRef]
113. Faridar A , Gamez N , Li D , Wang Y , Boradia R , Thome AD , et al. Low-dose interleukin-2 in patients with mild to moderate Alzheimer’s disease: A randomized clinical trial. Alzheimers Res Ther. 2025; 17( 1): 146. doi:10.1186/s13195-025-01791-x. [Google Scholar] [CrossRef]
114. Izzy S , Yahya T , Albastaki O , Abou-El-Hassan H , Aronchik M , Cao T , et al. Nasal anti-CD3 monoclonal antibody ameliorates traumatic brain injury, enhances microglial phagocytosis and reduces neuroinflammation via IL-10-dependent Treg-microglia crosstalk. Nat Neurosci. 2025; 28( 3): 499– 516. doi:10.1038/s41593-025-01877-7. [Google Scholar] [CrossRef]
115. U. S. National Library of Medicine . A Study of Intranasal Foralumab in Participants with Mild Cognitive Impairment due to Alzheimer’s Disease and Mild Alzheimer’s Disease (ClinicalTrials.gov Identifier: NCT06489548). 2025 [cited 2026 Jan 13]. Available from: https://clinicaltrials.gov/study/NCT06489548. [Google Scholar]
116. Tiziana Life Sciences plc . Phase II Clinical Updates for Foralumab in Alzheimer’s Disease. 2025 [cited 2026 Jan 13]. Available from: https://www.tizianalifesciences.com/drug-pipeline/intranasal-foralumab/. [Google Scholar]
117. Price BR , Sudduth TL , Weekman EM , Johnson S , Hawthorne D , Woolums A , et al. Therapeutic Trem2 activation ameliorates amyloid-beta deposition and improves cognition in the 5XFAD model of amyloid deposition. J Neuroinflammation. 2020; 17( 1): 238. doi:10.1186/s12974-020-01915-0. [Google Scholar] [CrossRef]
118. Wang S , Mustafa M , Yuede CM , Salazar SV , Kong P , Long H , et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J Exp Med. 2020; 217( 9): e20200785. doi:10.1084/jem.20200785. [Google Scholar] [CrossRef]
119. Long H , Simmons A , Mayorga A , Burgess B , Nguyen T , Budda B , et al. Preclinical and first-in-human evaluation of AL002, a novel TREM2 agonistic antibody for Alzheimer’s disease. Alzheimers Res Ther. 2024; 16( 1): 235. doi:10.1186/s13195-024-01599-1. [Google Scholar] [CrossRef]
120. Mayorga AJ , Burgess B , Nguyen T , Gao J , Salvadore G , Romano G . Baseline characteristics for INVOKE-2: A phase 2 randomized, double-blind, placebo-controlled study evaluating AL002 in early Alzheimer’s disease. Alzheimer’s Dement. 2024; 20: e095594. doi:10.1002/alz.095594. [Google Scholar] [CrossRef]
121. U. S. National Library of Medicine . Neurodegenerative Alzheimer’s Disease and Amyotrophic Lateral Sclerosis (NADALS) Basket Trial of Baricitinib (ClinicalTrials.gov Identifier: NCT05189106). 2023 [cited 2026 Jan 13]. Available from: https://clinicaltrials.gov/study/NCT05189106. [Google Scholar]
122. Alzheimer’s Research Forum . Baricitinib. n.d. [cited 2026 Jan 14]. Available from: https://www.alzforum.org/therapeutics/baricitinib. [Google Scholar]
123. Dempsey C , Rubio Araiz A , Bryson KJ , Finucane O , Larkin C , Mills EL , et al. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav Immun. 2017; 61: 306– 16. doi:10.1016/j.bbi.2016.12.014. [Google Scholar] [CrossRef]
124. Carreras I , Aytan N , Choi JK , Tognoni CM , Kowall NW , Jenkins BG , et al. Dual dose-dependent effects of fingolimod in a mouse model of Alzheimer’s disease. Sci Rep. 2019; 9( 1): 10972. doi:10.1038/s41598-019-47287-1. [Google Scholar] [CrossRef]
125. Wang MT , Hu ZC , Xiang Y , Zeng XQ , Fei ZC , Chen J , et al. Fingolimod ameliorates amyloid deposition and neurodegeneration in APP/PS1 mouse model of Alzheimer’s disease. J Prev Alzheimers Dis. 2025; 12( 7): 100131. doi:10.1016/j.tjpad.2025.100131. [Google Scholar] [CrossRef]
126. Calsolaro V , Edison P . Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016; 12( 6): 719– 32. doi:10.1016/j.jalz.2016.02.010. [Google Scholar] [CrossRef]
127. U. S. Food and Drug Administration . Tecfidera (Dimethyl Fumarate): Prescribing Information. 2023 [cited 2026 Jan 14]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2023/204063Orig1s029ltr.pdf. [Google Scholar]
128. Cui X , Zong S , Song W , Wang C , Liu Y , Zhang L , et al. Omaveloxolone ameliorates cognitive dysfunction in APP/PS1 mice by stabilizing the STAT3 pathway. Life Sci. 2023; 335: 122261. doi:10.1016/j.lfs.2023.122261. [Google Scholar] [CrossRef]
129. Parry-Jones AR , Stocking K , MacLeod MJ , Clarke B , Werring DJ , Muir KW , et al. Phase II randomised, placebo-controlled, clinical trial of interleukin-1 receptor antagonist in intracerebral haemorrhage: BLOcking the Cytokine IL-1 in ICH (BLOC-ICH). Eur Stroke J. 2023; 8( 3): 819– 27. doi:10.1177/23969873231185208. [Google Scholar] [CrossRef]
130. U. S. Food and Drug Administration . Arcalyst (Rilonacept): Prescribing Information. 2023 [cited 2026 Jan 26]. Available from: https://www.arcalysthcp.com/prescribing-information.pdf. [Google Scholar]
131. U. S. Food and Drug Administration . Ilaris (Canakinumab): Prescribing Information. 2023 [cited 2026 Jan 26]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/125319s107lbl.pdf. [Google Scholar]
132. Turner RS , Thomas RG , Craft S , van Dyck CH , Mintzer J , Reynolds BA , et al. Alzheimer’s Disease Cooperative Study. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology. 2015; 85( 16): 1383– 91. doi:10.1212/WNL.0000000000002035. [Google Scholar] [CrossRef]
133. Zhu CW , Grossman H , Neugroschl J , Parker S , Burden A , Luo X , et al. A randomized, double-blind, placebo-controlled trial of resveratrol with glucose and malate (RGM) to slow the progression of Alzheimer’s disease: A pilot study. Alzheimers Dement (N Y). 2018; 4: 609– 16. doi:10.1016/j.trci.2018.09.009. [Google Scholar] [CrossRef]
134. Patel S , Thornton A , Parmar MS . Resveratrol’s multifaceted potential in Alzheimer’s disease: Insights from preclinical and clinical evidence. Mol Neurobiol. 2025; 62( 12): 16229– 60. doi:10.1007/s12035-025-05234-4. [Google Scholar] [CrossRef]
135. Ringman JM , Frautschy SA , Teng E , Begum AN , Bardens J , Beigi M , et al. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res Ther. 2012; 4( 5): 43. doi:10.1186/alzrt146. [Google Scholar] [CrossRef]
136. Goozee K , Chatterjee P , Sohrabi HR , Martins R . Targeting preclinical stages of Alzheimer’s disease: A clinical trial to assess the efficacy of curcumin using brain biomarkers. Alzheimers Dement. 2016; 12( 7): 616. doi:10.1016/j.jalz.2016.06.1224. [Google Scholar] [CrossRef]
137. Chico L , Ienco EC , Bisordi C , Lo Gerfo A , Petrozzi L , Petrucci A , et al. Amyotrophic lateral sclerosis and oxidative stress: A double-blind therapeutic trial after curcumin supplementation. CNS Neurol Disord Drug Targets. 2018; 17( 10): 767– 79. doi:10.2174/1871527317666180720162029. [Google Scholar] [CrossRef]
138. George J . TREM2 as an evolving therapeutic target in Alzheimer’s disease. Neural Regen Res. 2023; 18( 12): 2680– 1. doi:10.4103/1673-5374.371360. [Google Scholar] [CrossRef]
139. Yuan P , Condello C , Keene CD , Wang Y , Bird TD , Paul SM , et al. TREM2 Haplodeficiency in mice and humans ımpairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. 2016; 90( 4): 724– 39. doi:10.1016/j.neuron.2016.05.003. [Google Scholar] [CrossRef]
140. Iguchi A , Takatori S , Kimura S , Muneto H , Wang K , Etani H , et al. INPP5D modulates TREM2 loss-of-function phenotypes in a β-amyloidosis mouse model. iScience. 2023; 26( 4): 106375. doi:10.1016/j.isci.2023.106375. [Google Scholar] [CrossRef]
141. Singhal T , Cicero S , Gale SA , Horan N , Dubey S , Marshall GA , et al. Dampening of microglial activation with nasal foralumab administration in moderate Alzheimer’s disease dementia. Clin Nucl Med. 2025; 50( 8): 756– 7. doi:10.1097/RLU.0000000000005955. [Google Scholar] [CrossRef]
142. Lopes JR , Zhang X , Mayrink J , Tatematsu BK , Guo L , LeServe DS , et al. Nasal administration of anti-CD3 monoclonal antibody ameliorates disease in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2023; 120( 37): e2309221120. doi:10.1073/pnas.2309221120. [Google Scholar] [CrossRef]
143. Campolo M , Casili G , Lanza M , Filippone A , Paterniti I , Cuzzocrea S , et al. Multiple mechanisms of dimethyl fumarate in amyloid β-induced neurotoxicity in human neuronal cells. J Cell Mol Med. 2018; 22( 2): 1081– 94. doi:10.1111/jcmm.13358. [Google Scholar] [CrossRef]
144. Izumi Y , Koyama Y . Nrf2-independent anti-inflammatory effects of dimethyl fumarate: Challenges and prospects in developing electrophilic Nrf2 activators for neurodegenerative diseases. Antioxidants. 2024; 13( 12): 1527. doi:10.3390/antiox13121527. [Google Scholar] [CrossRef]
145. Richardson PJ , Smith DP , de Giorgio A , Snetkov X , Almond-Thynne J , Cronin S , et al. Janus kinase inhibitors are potential therapeutics for amyotrophic lateral sclerosis. Transl Neurodegener. 2023; 12( 1): 47. doi:10.1186/s40035-023-00380-y. [Google Scholar] [CrossRef]
146. Hindam MO , Ahmed LA , El Sayed NS , Khattab M , Sallam NA . Repositioning of baricitinib for management of memory impairment in ovariectomized/D-galactose treated rats: A potential role of JAK2/STAT3-PI3K/AKT/mTOR signaling pathway. Life Sci. 2024; 351: 122838. doi:10.1016/j.lfs.2024.122838. [Google Scholar] [CrossRef]
147. Chen Y , Yang X , Li J , Luo H , Huang Q , Yang W , et al. A nasally administrated reactive oxygen species-responsive carrier-free gene delivery nanosystem for Alzheimer’s disease combination therapy. J Control Release. 2025; 381: 113604. doi:10.1016/j.jconrel.2025.113604. [Google Scholar] [CrossRef]
148. Pournajaf S , Dargahi L , Javan M , Pourgholami MH . Molecular Pharmacology and Novel Potential Therapeutic Applications of Fingolimod. Front Pharmacol. 2022; 13: 807639. doi:10.3389/fphar.2022.807639. [Google Scholar] [CrossRef]
149. Potenza RL , De Simone R , Armida M , Mazziotti V , Pèzzola A , Popoli P , et al. A disease-modifier drug in a mouse model of amyotrophic lateral sclerosis. Neurotherapeutics. 2016; 13( 4): 918– 27. doi:10.1007/s13311-016-0462-2. [Google Scholar] [CrossRef]
150. Berry JD , Paganoni S , Atassi N , Macklin EA , Goyal N , Rivner M , et al. Phase IIa trial of fingolimod for amyotrophic lateral sclerosis demonstrates acceptable acute safety and tolerability. Muscle Nerve. 2017; 56( 6): 1077– 84. doi:10.1002/mus.25733. [Google Scholar] [CrossRef]
151. Kubat Oktem E , Aydin B , Yazar M , Arga KY . Integrative analysis of motor neuron and microglial transcriptomes from SOD1G93A Mice models uncover potential drug treatments for ALS. J Mol Neurosci. 2022; 72( 11): 2360– 76. doi:10.1007/s12031-022-02071-1. [Google Scholar] [CrossRef]
152. Shokr MM . Rewiring brain immunity: Targeting microglial metabolism for neuroprotection in neurodegenerative disorders. Metab Brain Dis. 2025; 40( 8): 326. doi:10.1007/s11011-025-01739-y. [Google Scholar] [CrossRef]
153. Kanazawa M , Ninomiya I , Hatakeyama M , Takahashi T , Shimohata T . Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int J Mol Sci. 2017; 18( 10): 2135. doi:10.3390/ijms18102135. [Google Scholar] [CrossRef]
154. Song GJ , Suk K . Pharmacological modulation of functional phenotypes of microglia in neurodegenerative diseases. Front Aging Neurosci. 2017; 9: 139. doi:10.3389/fnagi.2017.00139. [Google Scholar] [CrossRef]
155. Li C , Ren J , Zhang M , Wang H , Yi F , Wu J , et al. The heterogeneity of microglial activation and its epigenetic and non-coding RNA regulations in the immunopathogenesis of neurodegenerative diseases. Cell Mol Life Sci. 2022; 79( 10): 511. doi:10.1007/s00018-022-04536-3. [Google Scholar] [CrossRef]
156. Xu YJ , Au NPB , Ma CHE . Functional and phenotypic diversity of microglia: Implication for microglia-based therapies for Alzheimer’s disease. Front Aging Neurosci. 2022; 14: 896852. doi:10.3389/fnagi.2022.896852. [Google Scholar] [CrossRef]
157. Krasemann S , Madore C , Cialic R , Baufeld C , Calcagno N , El Fatimy R , et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017; 47( 3): 566– 81.e9. doi:10.1016/j.immuni.2017.08.008. [Google Scholar] [CrossRef]
158. Vidovic N , Spittau B . Microglial transforming growth factor-β signaling in Alzheimer’s disease. Int J Mol Sci. 2024; 25( 6): 3090. doi:10.3390/ijms25063090. [Google Scholar] [CrossRef]
159. Parodi B , Rossi S , Morando S , Cordano C , Bragoni A , Motta C , et al. Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol. 2015; 130( 2): 279– 95. doi:10.1007/s00401-015-1422-3. [Google Scholar] [CrossRef]
160. Peng H , Li H , Sheehy A , Cullen P , Allaire N , Scannevin RH . Dimethyl fumarate alters microglia phenotype and protects neurons against proinflammatory toxic microenvironments. J Neuroimmunol. 2016; 299: 35– 44. doi:10.1016/j.jneuroim.2016.08.006. [Google Scholar] [CrossRef]
161. Hickman S , Izzy S , Sen P , Morsett L , El Khoury J . Microglia in neurodegeneration. Nat Neurosci. 2018; 21( 10): 1359– 69. doi:10.1038/s41593-018-0242-x. [Google Scholar] [CrossRef]
162. Guillot-Sestier MV , Town T . Let’s make microglia great again in neurodegenerative disorders. J Neural Transm. 2018; 125( 5): 751– 70. doi:10.1007/s00702-017-1792-x. [Google Scholar] [CrossRef]
163. Park J , Min JS , Kim B , Chae UB , Yun JW , Choi MS , et al. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-κB pathways. Neurosci Lett. 2015; 584: 191– 6. doi:10.1016/j.neulet.2014.10.016. [Google Scholar] [CrossRef]
164. Takahashi K , Rochford CD , Neumann H . Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005; 201( 4): 647– 57. doi:10.1084/jem.20041611. [Google Scholar] [CrossRef]
165. Zheng H , Jia L , Liu CC , Rong Z , Zhong L , Yang L , et al. TREM2 promotes microglial survival by activating Wnt/β-catenin pathway. J Neurosci. 2017; 37( 7): 1772– 84. doi:10.1523/JNEUROSCI.2459-16.2017. [Google Scholar] [CrossRef]
166. Yu IC , Paraiso HC , Kuo P , Scofield BA , Sweazey RD , Chang FL , et al. Functional Nrf2 restrains inflammatory and transcriptional phenotypes in microglia and its deficiency recapitulates the aging phenotype. J Immunol. 2019; 202( 1 Supplement): 185.17. doi:10.4049/jimmunol.202.Supp.185.17. [Google Scholar] [CrossRef]
167. Chu CT , Uruno A , Katsuoka F , Yamamoto M . Role of NRF2 in pathogenesis of Alzheimer’s disease. Antioxidants. 2024; 13( 12): 1529. doi:10.3390/antiox13121529. [Google Scholar] [CrossRef]
168. Coll RC , Schroder K , Pelegrín P . NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci. 2022; 43( 8): 653– 68. doi:10.1016/j.tips.2022.04.003. [Google Scholar] [CrossRef]
169. Ivanenkov YA , Balakin KV , Lavrovsky Y . Small molecule inhibitors of NF-kB and JAK/STAT signal transduction pathways as promising anti-inflammatory therapeutics. Mini Rev Med Chem. 2011; 11( 1): 55– 78. doi:10.2174/138955711793564079. [Google Scholar] [CrossRef]
170. Nizami S , Hall-Roberts H , Warrier S , Cowley SA , Di Daniel E . Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br J Pharmacol. 2019; 176( 18): 3515– 32. doi:10.1111/bph.14618. [Google Scholar] [CrossRef]
171. Yi YS , Kim HG , Kim JH , Yang WS , Kim E , Park JG , et al. Syk promotes phagocytosis by inducing reactive oxygen species generation and suppressing SOCS1 in macrophage-mediated inflammatory responses. Int J Immunopathol Pharmacol. 2022; 36: 3946320221133018. doi:10.1177/03946320221133018. [Google Scholar] [CrossRef]
172. Chen T , Shen L , Yu J , Wan H , Guo A , Chen J , et al. Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell. 2011; 10( 5): 908– 11. doi:10.1111/j.1474-9726.2011.00722.x. [Google Scholar] [CrossRef]
173. Abdi M , Pasbakhsh P , Shabani M , Nekoonam S , Sadeghi A , Fathi F , et al. Metformin therapy attenuates pro-inflammatory microglia by ınhibiting NF-κB in cuprizone demyelinating mouse model of multiple sclerosis. Neurotox Res. 2021; 39( 6): 1732– 46. doi:10.1007/s12640-021-00417-y. [Google Scholar] [CrossRef]
174. Yang B , Pan J , Zhang XN , Wang H , He L , Rong X , et al. NRF2 activation suppresses motor neuron ferroptosis induced by the SOD1G93A mutation and exerts neuroprotection in amyotrophic lateral sclerosis. Neurobiol Dis. 2023; 184: 106210. doi:10.1016/j.nbd.2023.106210. [Google Scholar] [CrossRef]
175. Liu Z , Jia J . Omaveloxolone ameliorates cognitive deficits by inhibiting apoptosis and neuroinflammation in APP/PS1 mice. Mol Neurobiol. 2025; 62( 2): 2191– 202. doi:10.1007/s12035-024-04361-8. [Google Scholar] [CrossRef]
176. Șerban M , Toader C , Covache-Busuioc RA . The redox revolution in brain medicine: Targeting oxidative stress with AI, multi-omics and mitochondrial therapies for the precision eradication of neurodegeneration. Int J Mol Sci. 2025; 26( 15): 7498. doi:10.3390/ijms26157498. [Google Scholar] [CrossRef]
177. Sharkus R , Thakkar R , Kolson DL , Constantinescu CS . Dimethyl fumarate as potential treatment for Alzheimer’s Disease: Rationale and clinical trial design. Biomedicines. 2023; 11( 5): 1387. doi:10.3390/biomedicines11051387. [Google Scholar] [CrossRef]
178. Cabral JE , Wu A , Zhou H , Pham MA , Lin S , McNulty R . Targeting the NLRP3 inflammasome for inflammatory disease therapy. Trends Pharmacol Sci. 2025; 46( 6): 503– 19. doi:10.1016/j.tips.2025.04.007. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools