
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Melanoma-derived extracellular vesicles transfer proangiogenic factors
1 Department of Glycoconjugate Biochemistry, Faculty of Biology, Institute of Zoology and Biomedical Research, Jagiellonian University, Krakow, 30-387, Poland
2 Doctoral School of Exact and Natural Sciences, Jagiellonian University, Krakow, 30-348, Poland
* Corresponding Authors: MAGDALENA SURMAN. Email: ; MAłGORZATA PRZYBYłO. Email:
# Authors contributed equally to the manuscript
Oncology Research 2025, 33(2), 245-262. https://doi.org/10.32604/or.2024.055449
Received 27 June 2024; Accepted 27 September 2024; Issue published 16 January 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Angiogenesis, the expansion of pre-existing vascular networks, is crucial for normal organ growth and tissue repair, but is also involved in various pathologies, including inflammation, ischemia, diabetes, and cancer. In solid tumors, angiogenesis supports growth, nutrient delivery, waste removal, and metastasis. Tumors can induce angiogenesis through proangiogenic factors including VEGF, FGF-2, PDGF, angiopoietins, HGF, TNF, IL-6, SCF, tryptase, and chymase. This balance is disrupted in tumors, and extracellular vesicles (EVs) contribute to this by transferring proangiogenic factors and increasing their expression in endothelial cells (ECs). Malignant melanoma, a particular type of skin cancer, accounts for only 1% of skin cancer cases but more than 75% of deaths. Its incidence has risen significantly, with a 40% increase between 2012 and 2022, especially in fair-skinned populations. Advanced metastatic stages have a high mortality due to delayed diagnosis. This review examines the molecular basis of angiogenesis in melanoma, focusing on melanoma-derived EVs and their possible use in new antiangiogenic therapies.Keywords
Angiogenesis is a multistep process that leads to the expansion of pre-existing vascular and microvascular networks in all organs and tissues [1]. It is essential for normal organ growth and is required for the repair of any damaged tissue. However, aberrant angiogenesis can be observed in a variety of pathologies, including inflammation, ischemia, diabetes, varicose veins, hemangiomas, aneurysms, and many others. Angiogenesis plays a particularly important role in cancer, as solid tumors larger than a few millimeters in size require a constant blood supply. Tumor vasculature enables oxygen and nutrient delivery to growing tumors, removal of metabolic wastes, and provides a route for local and distant metastasis [1].
Most tumors are also capable of producing various molecular signals to induce or enhance angiogenesis. Main proangiogenic factors can be divided into two groups, i.e., 1) classical factors such as angiopoietins (ANGPTs), fibroblast growth factor-2 (FGF-2), hepatocyte growth factor (HGF), interleukin-6 (IL-6), platelet-derived growth factor (PDGF), tumor necrosis factor (TNF), and vascular endothelial growth factor (VEGF), as well as 2) non-classical factors such as chymase, stem cell factor (SCF), and tryptase [2–4]. The above-mentioned and other proangiogenic factors act in balance with antiangiogenic molecules under physiological conditions.
Disruption of this homeostasis is observed in the tumor microenvironment (TME). The secretion and intercellular transfer of proangiogenic factors are increased, causing alternations to the structure of the extracellular matrix (ECM) and expansion of tumor vasculature. The factors contributing to tumor angiogenesis include among others extracellular vesicles (EVs)–nanosized, phospholipid bilayer-enclosed particles, involved in the horizontal transfer of specific molecular cargo between almost all cell types in the human body. EVs have already been shown to carry several proangiogenic factors, including VEGF, matrix metalloproteinases (MMPs), and their endogenous activator CD147, PDGF, microRNAs, and lncRNAs as well as up-regulating their expression in ECs [5,6], thus facilitating interactions between tumor and ECs or other cells involved in angiogenesis.
Malignant melanoma is one of the most aggressive cancers, and although it is responsible for only 1% of skin cancer cases, it accounts for more than 75% of skin cancer-related mortality [7,8]. The last few decades have brought a decline in the incidence and mortality rates of most cancers, but nevertheless, the incidence of melanoma has increased, especially in fair-skinned populations. Between 2012 and 2022, there was more than 40% increase in melanoma cases per year [9]. A particularly high mortality rate is associated with advanced metastatic stages of the disease (Stages III and IV), mainly due to delayed diagnosis. Intravasation of tumor cells is a key step to subsequent metastasis and disease containment at this stage significantly improves patient outcomes.
Therefore, in this review, we cover the molecular basis behind angiogenesis in melanoma. We focus on the role of melanoma-derived EVs in this process and on their possible use in new antiangiogenic therapies.
Angiogenesis is central to the progression of solid tumors, and melanoma is no exception. The development of primary cutaneous melanoma is usually divided into two growth phases, radial phase and vertical phase. In the radial phase, melanoma forms an irregular plaque, and tumor cells cannot invade the dermis. In the vertical phase, the lesion grows vertically into deeper parts of the dermis. During the vertical growth phase, angiogenesis is crucial for tumor growth and metastasis, which is strictly associated with the invasion of blood or lymphatic vessels. Melanoma metastatic sites are most commonly observed in the bones, lungs, liver, and brain [10].
The uncontrolled proliferation of melanoma cells leads to increased energy demand, which manifests as a drastic decrease in oxygen in TME and the development of hypoxia. Hypoxia is present in 90% of solid tumors and occurs in tissues more than 100–200 µM away from the functional blood supply [11]. In response to hypoxia the hypoxia-inducible factor (HIF) pathway is activated (Fig. 1). HIF is composed of an alpha subunit (HIF-α) and a beta subunit (HIF-β). HIF-α is the main transcription regulator of the developmental response to hypoxia [12]. The HIF-α subunits (HIF-1α, HIF-2α, and HIF-3α) are stabilized by enzymes that are oxygen sensors, i.e., factor inhibiting HIF-1 (FIH-1) and prolyl hydroxylase domain (PHD) [13,14]. In an oxygen-available environment, PHD and FIH-1 hydroxylate HIF-α, and hydroxylated HIF-α are then tagged by Von Hippel–Lindau tumor suppressor with E3 ubiquitin ligase activity. Ubiquitinated HIF-α is then degraded in proteasomes [15]. Reduced oxygen availability leads to inhibition of PHD and FIH-1 function and stops degradation of HIF-α subunits, which are transported to the nucleus and dimerized with HIF-β subunits and act as a transcription factor [16].
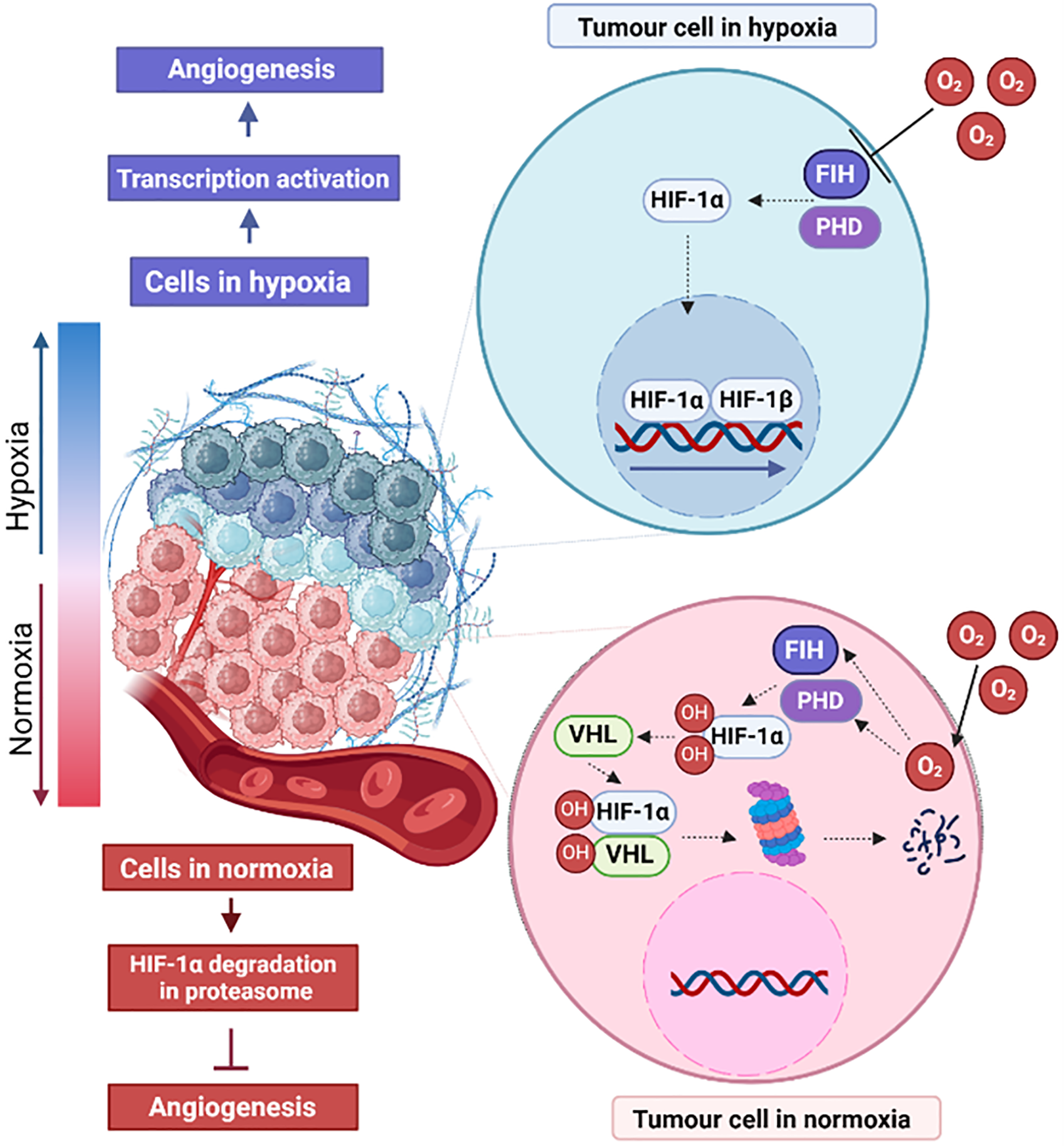
Figure 1: Effects of normoxia and hypoxia on angiogenesis in tumor cells. HIF-1α-hypoxia-inducible factor 1 subunit α, HIF-1β-hypoxia-inducible factor 1 subunit β, VHL-Von Hippel–Lindau tumor suppressor, PHD-prolyl hydroxylase domain enzyme, FIH-factor-inhibiting HIF. The figure was prepared under the license in BioRender: Scientific Image and Illustration Software.
The HIF-1α and the nuclear factor ĸB (NF-ĸB) pathways interact with other proangiogenic factors, such as cyclooxygenase-2 (COX-2), cytokine-inducible nitric oxide synthase (iNOS), and stromal cell-derived factor 1(SDF-1) VEGF, and VEGF receptor (VEGFR) [17]. HIF-α is mainly involved in the recruitment of bone marrow endothelial progenitor cells (EPCs), which later differentiate into ECs via the VEGF pathway, thereby stimulating vascularization [16].
Angiogenesis is preceded by vasculogenesis, de novo formation of a primitive vascular network (mainly during embryogenesis) from bone marrow-derived EPCs. Subsequently, this pre-existing vasculature, made of differentiated ECs, takes part in the formation and expansion of new blood vessels in angiogenesis. There are several mechanisms of blood vessel formation [18–20]. The vessels formed by ECs are then strengthened by pericytes and smooth muscle cells, which enable perfusion. Blood vessels can grow both by sprouting and by a non-sprouting mechanism of intussusceptive microvascular growth (IMG). In sprouting angiogenesis, the basement membrane undergoes rearrangement at the site of the dilated peritumoral postcapillary venule in proximity to the angiogenic stimulus. The ECs then relocate to the connective tissue to form a solid cord, and the migrating front leads to lumen formation. Non-sprouting angiogenesis is a recovery adaptation of the existing microvascular network. Unlike traditional angiogenesis, which depends on the rapid proliferation of ECs, this process results from the reorganization of existing ECs and the incorporation of EPCs. Intussusceptive angiogenesis is related to the presence of the intussusceptive pillar, a transvascular tissue bridge of 1–5 μm in length that spans the vessel lumen. Therefore, the vascular network expands by the insertion of pillars.
Proangiogenic and antiangiogenic factors in melanoma
There are many proangiogenic factors released by melanoma cells (Fig. 2), which form a complex intra-and intercellular signaling network [4]. Some of these factors including VEGF, PDGF, or ANGPTs, generally stimulate melanoma-related angiogenesis by directly affecting the basic functions (such as proliferation or migration) of ECs. In contrast, factors such as IL-8, metalloproteinases (MMPs), or integrins are more indirect mediators, although still important contributors to the development of tumor vasculature. They usually act as cofactors for the factors from the first group or promote their secretion. They may be also involved in ECM remodeling required for angiogenesis, without direct interaction with ECs [4]. The specific mechanisms of action of the main proangiogenic factor identified in melanoma are described in the following subsections.
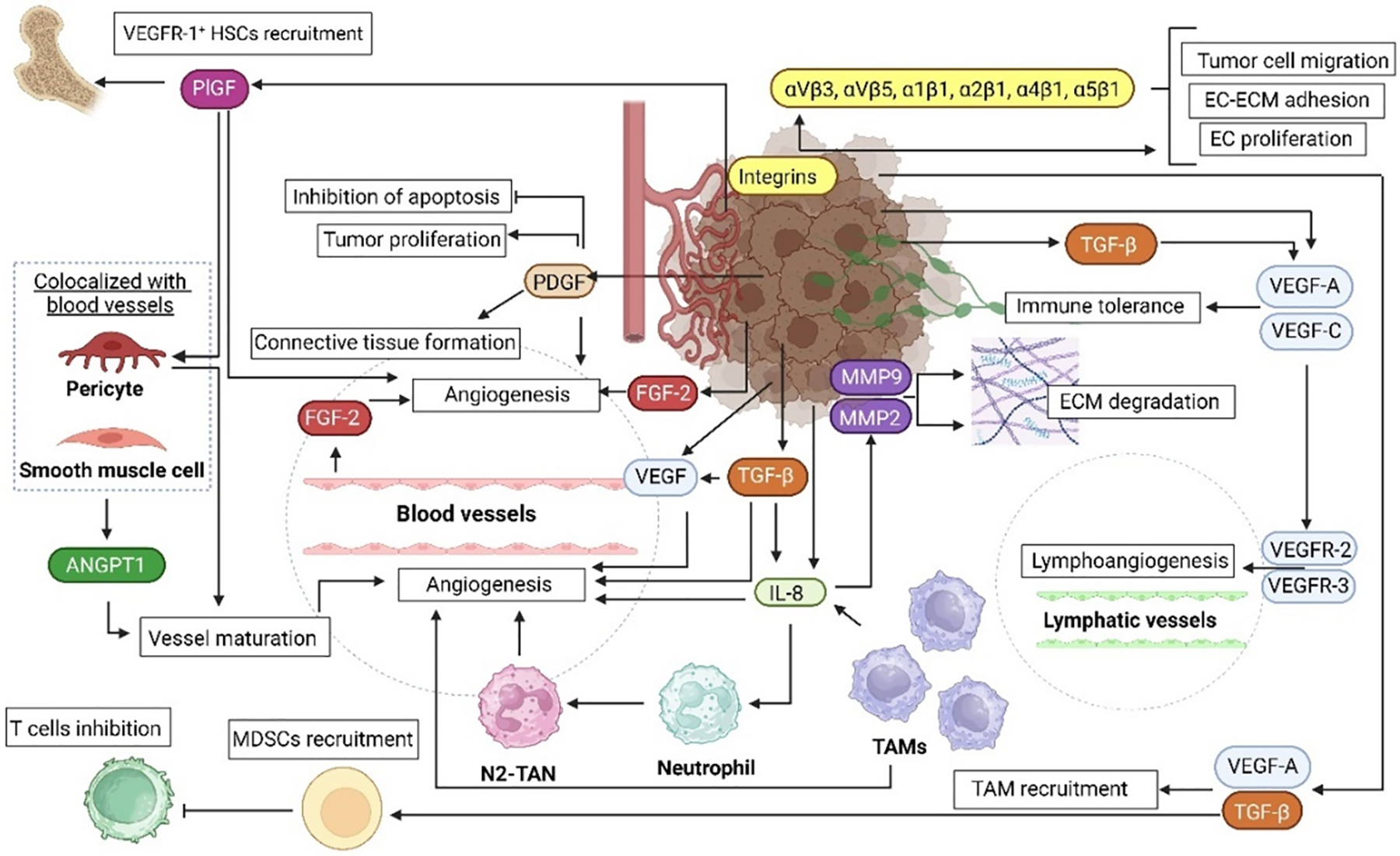
Figure 2: Schematic summary of the role of proangiogenic factors in melanoma. Melanoma microenvironment consists of different cell types, such as immune system cells (TAMs, neutrophils, T cells) and endothelial cells. These cells cooperate in multiple processes involved in melanoma progression, one of which is angiogenesis. The coexistence of the cells forms paracrine loops involved in ECM degradation, lymphoangiogenesis, and importantly the formation of new blood vessel and their maturation. ANGPT–angiopoietin, EC–endothelial cell, ECM–extracellular matrix, FGF–fibroblast growth factor, HSC–hematopoietic stem cell, IL-8–interleukin 8, MDSC–myeloid-derived suppressor cell, MMP–matrix metalloproteinase, PDGF–platelet-derived growth factor, PlGF–placental growth factor, TAM–tumor-associated macrophages, TGF-β–transforming growth factor β, VEGF(R)–vascular endothelial growth factor (receptor). The figure was prepared under the license in BioRender: Scientific Image and Illustration Software.
Vascular endothelial growth factor (VEGF)
VEGF is a signaling protein produced by various types of cells. The human VEGF family consists of VEGF-A, B, C, D, and E. All VEGF isoforms elicit a response through cell surface tyrosine kinase receptors. Binding of VEGF results in their dimerization and activation by transphosphorylation [21]. VEGF has been shown to be the survival factor for ECs in vivo and in vitro [22]. ECs of newly formed vessels functionally rely on VEGF, in contrast to established vessels within tumors. The loss of VEGF dependence may be explained by the coverage of ECs by pericytes [23].
VEGF also contributes to the horizontal/vertical growth phase transition in melanoma [24]. Moreover, strong reactivity in anti-VEGF immunohistochemical staining of melanoma specimens and increased microvascular density correlate positively with the formation of tumors of larger thicknesses (>3.6 mm). This supports the hypothesis that VEGF is involved in increasing vessel diameter and IMG in melanoma [25].
Studies on melanoma cell lines also revealed that cells with low VEGF expression can be stimulated to increase VEGF secretion by culture in hypoxic conditions [26]. Clinical studies have also shown a positive correlation between VEGF levels and Breslow scale depth and with Clark scale levels [27].
Furthermore, VEGF-A promotes the proliferation of VEGFR-2-positive cells in lymphatic vessels and metastasis to sentinel and distant lymph nodes [28]. VEGF-C also induces tumor lymphangiogenesis and enhances melanoma metastasis to lymph nodes by binding to VEGFR-3 [29]. VEGF-C expression, as well as intratumoral lymphatic vessel density (LVD), peritumoral LVD, melanoma thickness, and Clark level are good predictors of lymph node metastasis in melanoma [30]. Similarly, VEGF-C/D expression in melanoma lymph node metastases was higher than in non-metastatic melanomas [31]. VEGF-C also enhances the transport of tumor cells to the draining lymph node and their exposure to immune cells. In murine melanoma, VEGF-C promoted immune tolerance [32]. Finally, VEGF-B was found to inhibit angiogenesis. The mechanism of action was based on the FGF-B binding to FGFR1, and subsequent FGFR1/VEGFR1 complex formation, which blocked Erk activation by FGF-B [33].
Platelet-derived growth factor (PDGF)
PDGF glycoprotein mainly exists in three variants, i.e., PDGF-AA consisting of two A subunits, PDGF-BB consisting of two B subunits, and PDGF-AB heterodimer. In the early 2000s, two additional family members, PDGF-C and PDGF-D, were identified as novel ligands for PDGF receptors (PDGFRs) [34]. Primary and metastatic melanoma is characterized by overexpression of two PDGFRs, namely PDGFR-α and PDGFR-β, compared to normal skin [35]. PDGF is secreted by melanoma cell lines and tumors, stimulating the development of new blood vessels [35]. Moreover, it inhibits melanoma apoptosis, promotes cell cycle progression enhances melanoma cell survival [36], and exhibits mitogenic properties for melanoma cells by stimulating their proliferation through MAPK/ERK and PI3K/Akt pathways.
Mice inoculated with PDGF-BB-transfected B16 melanoma cells showed an increased pericyte coverage of tumor blood vessels [37]. However, susceptibility-contrast Magnetic Resonance Imaging (MRI) showed a significant reduction in the vessel size index in tumors formed by B16 cells with PDGF overexpression [37]. Similarly, PDGFR-α overexpression abrogated the growth of some melanoma tumors [38]. Additionally, tumor-derived PDGF-BB dimer may mediate connective tissue stroma formation as mice inoculated with WM9 melanoma cells lacking PDGF-B subunit expression showed highly necrotic tumors with narrow lumen blood vessels and lack of connective tissue. Tumors formed by WM9 cells with PDGF-B subunit overexpression had properly developed vessels and connective tissue, which resulted in a lack of necrosis [39].
Human IL-8 is currently called chemokine (C-X-C motif) ligand 8 (CXCL8), and its receptors, previously known as IL8Rα and IL8Rβ, are now named CXCR1 and CXCR2, respectively [40]. In melanoma, CXCR2 expression is increased compared to neutrophils [41]. This results in increased binding of IL-8 to the tumor instead of immune cells. Normal melanocytes do not produce IL-8 unless stimulated. In melanoma, IL-8 mRNA is consistently expressed and is correlated with the tumor’s ability to metastasize [42]. However, IL-8 mRNA has also been observed in non-metastatic melanoma [38].
IL-8 is released by macrophages, epithelial or ECs and attracts neutrophils in infections and injuries. This results not only in the removal of pathogens, but also in enhanced angiogenesis, and the synthesis of MMPs [43,44]. Dermal microvascular ECs express IL8Rs, so IL-8 regulates MMP-2/-9 secretion and subsequent angiogenesis [45]. IL-8 is also a target of the nuclear factor of activated T cells, i.e., NFAT1 and NFATC2. NFATs are involved in immune response, but also in melanoma progression and metastasis. NFAT1 binds to the IL-8 promoter and increases IL-8 transcription, thereby promoting tumor development, growth, and dissemination [46].
Tumor-associated neutrophils (TANs) have anti-or pro-tumor phenotypes, N1-TANs, and N2-TANs, respectively [47]. IL-8 is one of the major factors in the N1-/N2-TANs balance, suppressing antitumor immunity by recruiting N2-TANs. TANs can produce IL-8, further stimulating neutrophil migration, vessel formation, and tumor growth [48]. Also, tumor-associated macrophages (TAMs) are key effectors in tumor angiogenesis, especially macrophage-derived angiogenesis and tumor invasiveness through processes mediated by IL-8 [49].
Fibroblast growth factor-2 (FGF-2)
FGF-2, also known as the basic fibroblast growth factor (bFGF), is a cytokine that binds to the fibroblast growth factor receptor (FGFR) [50,51]. FGF-2 is produced by melanoma cells and ECs [52]. In tumors, FGF-2 through FGFR2 stimulates the proliferation of pericytes and activates PDGFRβ signaling required for pericyte migration to the angiogenesis site [53]. During angiogenesis in melanoma, FGF-2 cooperates with heparinase, which enzymatically cleaves the glycosaminoglycan chains of heparan sulfate proteoglycans, which act as a receptor or coreceptor for FGFR [54].
Abnormal expression of FGF-2 (along with FGF-18) and elevated FGFR1 and FGFR3 characterize primary melanoma vs. healthy skin [55]. Also, analyses of the FGFR4 Arg388 polymorphism in 185 melanoma patients identified Arg388 allele in 45% of patients and was associated with tumor size and high microvascular density [56].
Four ANGPTs are known: ANGPT1, ANGPT2, ANGPTL3, and ANGPT4 [57]. ANGPT1 is secreted by pericytes and vascular smooth muscle cells. It is a key factor in the blood vessel maturation, adhesion, and migration of ECs. Its proangiogenic activity is induced by binding to the angiopoietin-1 receptor (Tie-2), which increases vessel quiescence and inhibits vascular permeability. ANGPT1 is significantly overexpressed in the vasculature of most tumors, and in ANGPT-1-deficient mice, MT-ret and B16F10 melanomas grow more slowly [58]. ANGPT2 is released by ECs and may act as a Tie-2 antagonist, leading to temporary disruption of existing blood vessels required for their further development, i.e., angiogenesis [57,59]. The balance between ANGPT1 and ANGPT2 undergoes a proangiogenic shift in melanoma. High levels of ANGPT2 compared to ANGPT1 positively correlate with tumor vascularity, tumor growth, and poorer prognosis [60].
Transforming growth factor β (TGF-β)
TGF-β is a class of cytokines with multiple biological functions. The TGF-β family includes a variety of molecular subtypes, among which TGF-β1, -β2, and -β3 are most extensively studied to date. Unlike normal melanocytes, melanoma cells not only escape cell cycle arrest induced by TGF-β but also produce it and respond to it at the gene level [61]. TGF-β1 is produced by melanocytes and melanoma tumor cells. On the other hand, TGF-β2 and -β3 are expressed heterogeneously only in nevi and melanomas, and their expression increases during tumor progression [62].
TGF-β is involved in the formation of peri-tumoral blood vessels by stimulating the secretion of IL-8 and VEGF-A. TGF-β also activates the migration of ECs to sites of angiogenesis within the tumor niche [61]. Endoglin (CD105) is a TGF-β receptor binding TGF-β1 and -β3, but not TGF-β2 [63]. In ECs, endoglin is crucial for angiogenesis since it facilitates the binding of TGF-β family members to activin receptor-like Kinase 1 (ALK 1). Endoglin expression in melanoma indicates angiogenesis [64].
Moreover, two bone morphogenetic proteins (BMPs) from the TGF-β family, namely BMP-4 and BMP-7, are frequently overexpressed in melanoma. Importantly, downregulated expression of BMP-4 in melanoma cells correlated with their diminished proangiogenic paracrine activity [65,66].
Placental growth factor (PlGF)
Melanoma cells express two PIGF isoforms, namely PlGF-1 and PlGF-2, which bind to neuropilin-1 and neuropilin-2 receptors on ECs [67]. In addition, PlGF forms heterodimers with VEGF, indirectly interacting with VEGFR-2 on ECs [68,69]. PlGF also enhances blood vessel maturation by interacting with VEGFR-1-positive pericytes [68]. Moreover, PlGF contributed to increased vessel branching, size, and stability in vivo [70]. Finally, a comparison of PlGF levels between patients with metastatic melanoma and healthy control revealed 20-fold higher plasma PlGF levels in patients [71].
Matrix metalloproteinases (MMPs)
MMPs are a group of more than 20 Zn2+-dependent endogenous peptidases that participate in wound healing, tissue remodeling, and angiogenesis [72,73]. MMPs catalyze the degradation of collagen, gelatin, elastin, fibronectin, and laminin, which are key ECM components. As a consequence, their activity supports tumor metastasis, allowing tumor cell migration from primary sites to metastatic niches [74].
All MMPs expressed in melanoma cells directly or indirectly participate in angiogenesis [75,76]. IL-8 enhances the activity of MMP-2 secreted by melanoma cells, which in turn promote melanoma invasion [77]. Moreover, through interaction between membrane matrix metalloproteinase type 1, MMP-2, and laminin-5γ2 chain fragments melanoma cells tend to display vasculogenic mimicry, the phenomenon in which tumor cells mimic EC activity to participate in neovascularization and the formation of a matrix-rich meshwork [78].
In melanoma cells, MMP-9 colocalizes with CD44, this interaction promotes the proteolytic activity of MMP-9 against type IV collagen [79]. ECM degradation by MMP-9 induces secretion of FGF and VEGF [80]. Moreover, in melanoma cells, the rapamycin-insensitive companion of mTOR complex 2 (Rictor-mTORC2) can phosphorylate AKT, causing overexpression of MMP-2 and MMP-9 and microvessel formation [81]. However, MMP-9 has been shown to be expressed only during the horizontal but not vertical growth phase [82]. Malignant melanoma cells express not only MMP-2 and 9, but also MMP-1, 13, and 14, and their inhibitors such as TIMP-1, 2, and 3 [75,76].
The protein family of integrins consists of heterodimeric (α and β subunits with non-covalent linkage) transmembrane cell surface receptors involved in cell-cell and cell-ECM adhesion. Integrin signaling cascades also modulate cell proliferation and survival, as shown, for example, in the focal adhesion kinase (FAK), Rho GTPase, MAPK/ERK, and PI3K/Akt/mTOR pathways. After the transition from the primary to metastatic phase melanoma cells overexpress chosen integrins, namely αvβ3, αvβ5, α2β1, α4β1 α1β1, and α5β1 integrins [60].
A major part of research concerning the proangiogenic role of integrins was related to αvβ3 integrin. Integrin αvβ3 is a classic vitronectin receptor, often overexpressed in developing blood vessels and required for angiogenesis. The β3 integrin subunit expressed by ECs can undergo phosphorylation via the VEGF/c-Src pathway, and the phosphorylated β3 integrin subunit promotes activation of VEGFR-2. Blocking αvβ3 integrin with antibodies or low-molecular-weight antagonists, (e.g., arginyl glycyl aspartic acid (RGD) mimetics) inhibited angiogenesis in in vitro and in vivo models [83,84], including tumor angiogenesis in melanoma. Knockout of αvβ3 integrin in mouse melanoma tumors led to reduced tumor growth and microvessel density [84,85]. Also, conditioned media from αvβ3 integrin-expressing and αvβ3 integrin-non-expressing melanoma cell cultures were collected and added to HUVEC cells. ECs treated with media deprived of αvβ3 integrin showed a significantly lower proliferation rate in the MTT assay, suggesting that αvβ3 integrin present in the melanoma secretome has a measurable functional effect on ECs [85]. Moreover, melanoma cells co-expressing α2bβ3 and αvβ3 integrins were shown to also have an increased expression of FGF-2 [86].
Another integrin, αvβ5 integrin, is involved in the regulation of neuropilin 1 (NRP-1)-dependent angiogenic pathways in melanoma. Inhibition of αvβ5 integrin prevents the formation of NRP-1/VEGF-A complexes and abrogates angiogenesis [82]. In addition, the β1 integrin subunit was found to be required for melanoma cell adhesion to ECs [87], mainly through the FAK/paxillin pathway, and for the extravasation of melanoma cells at metastasis sites (mainly in liver and lung) [88,89]. Also, α5β1 integrin modulates ANG-1-dependent angiogenesis through interaction with Tie-2 [90].
Interest in EVs is constantly increasing due to their enormous diagnostic and therapeutic potential. EVs can be isolated from body fluids (blood, breast milk, cerebrospinal fluid, saliva, sperm, urine,), and conditioned culture media [91]. Within EVs, three subpopulations of vesicular structures are usually distinguished, i.e., exosomes, ectosomes (also known as microvesicles), and apoptotic bodies (Fig. 3). The largest EVs are apoptotic bodies (ABs) (1000 to 5000 nm in diameter), which are released by cell shrinkage during apoptotic death. Biogenesis of ectosomes involves cell membrane budding caused by rearrangements in the cytoskeleton. MVs are also released constitutively, and an increase in their production by cells is usually associated with the action of various stressors, such as nutrient deprivation or increased Ca2+ levels. The diameter of MVs varies between 100–1000 nm. Exosomes are the smallest EVs (30 to 100 nm in diameter). Exosome biogenesis is a multistep process, in which endosomes are involved. After invagination of the endosome membrane intraluminal vesicles (ILVs) are formed. From this point on, endosomes transporting ILVs are called multivesicular bodies (MVBs). Once MVBs fuse with the cell membrane, exosomes are released to the extracellular space [92,93].
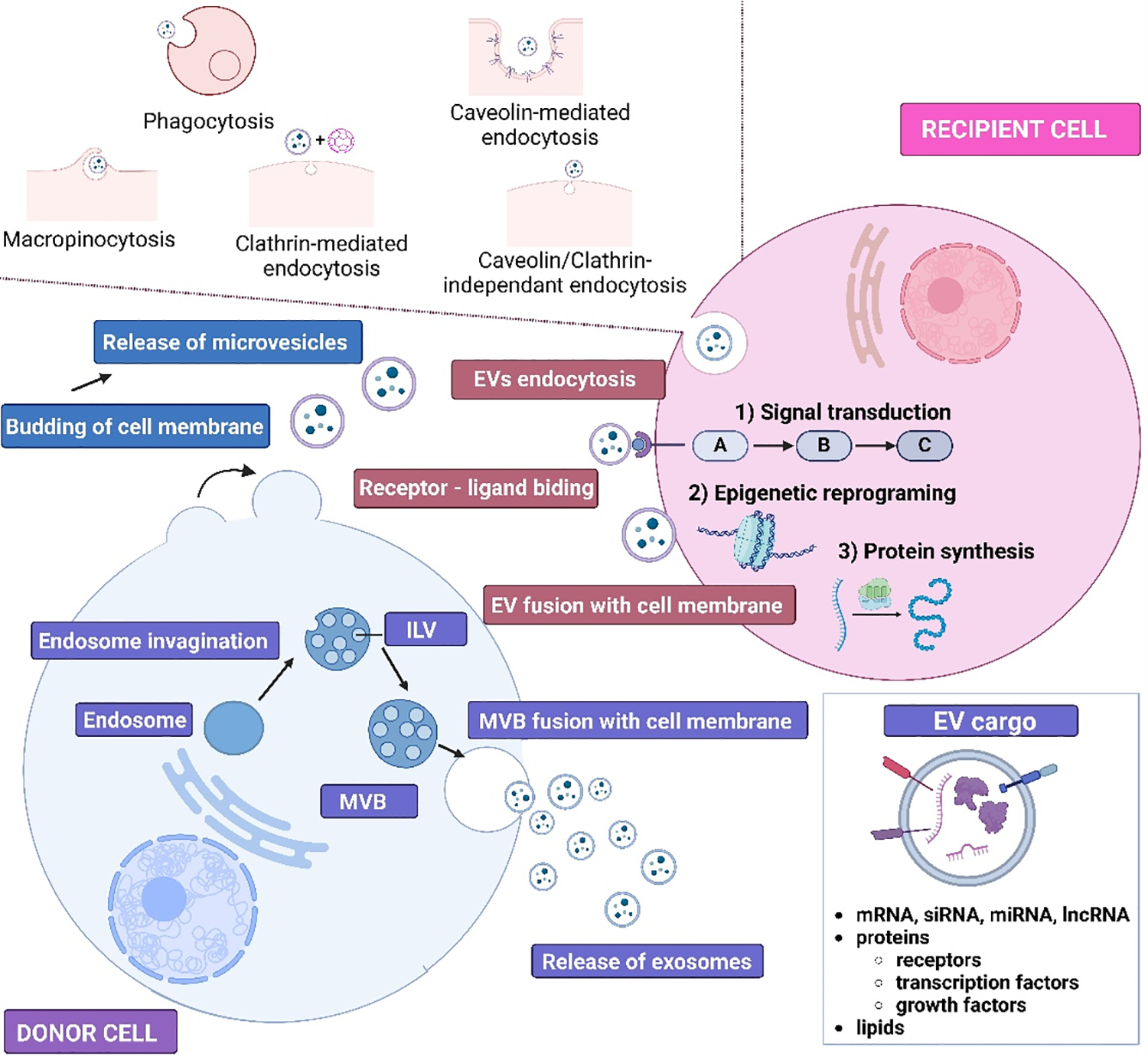
Figure 3: Schematic presentation of exosomes and microvesicles biogenesis and the ways of cargo transmission to recipient cells. ILV–Intraluminal vesicle, MVB–multivesicular body, EVs–extracellular vesicles, mRNA–messenger RNA, siRNA–small interfering RNA, miRNA–microRNA, lncRNA–long non-coding RNA. The figure was prepared under the license in BioRender: Scientific Image and Illustration Software.
However, the terminology for classifying EVs, including “exosomes”, “microvesicles/ectosomes” and “apoptotic bodies”, should be used with caution in accordance with Minimal information for studies of extracellular vesicles (MISEV2023) guidelines [94]. Most isolation methods (e.g., filtration or differential ultracentrifugation) separate EVs according to their size and density, whereas the terminology described here relates more strictly to their biogenesis. The size ranges (30–100 nm and 100–1000 nm) are highly arbitrary–exosomes and ectosomes probably overlap in size, especially around 100 ± 50 nm. Therefore, terms such as “small” (<200 nm) and “large” (>200 nm) have been commonly used to denote EV populations over the past few years, unless the cellular origin (endosomal vs. outer cell membrane) can be clearly demonstrated for an isolated sample. Thus, while different terminologies can still be used, researchers should be aware of their limitations and aim to characterize the EVs under study as clearly as possible [94].
When EVs were first observed, no role was assigned to them, as they were considered a type of cellular waste. It was later discovered that they participate in cellular communication and are involved in various physiological and pathological processes, e.g., autoimmune [95] and neurodegenerative diseases [96], processes associated with transplanted organ rejection [97], and cancer [98]. In carcinogenesis, EVs facilitate drug resistance, angiogenesis, epithelial-mesenchymal transition, invasion, migration, escape from apoptosis, and both pro-and anti-tumor stimulation of the immune system. All of these processes may be modified by the cargo transported by EVs, including metabolites, active forms of lipids, proteins (e.g., transcription factors, receptors, growth factors), and a broad panel of nucleic acids, including mRNAs, lncRNAs, and miRNAs [99].
The EV cargo largely mirrors the state of the cell that released them. The molecular information contained in EVs can be transmitted to the recipient cells in several ways as shown in Fig. 3 [100]. EVs can interact with recipient cells through various types of endocytosis, such as caveolin- or clathrin-mediated endocytosis, micropinocytosis, lipid raft-mediated endocytosis, or phagocytosis. EVs can also directly fuse with the membrane of recipient cell. Finally, the molecular signal can be transduced via receptor-ligand interaction. Therefore, the interaction between EVs and recipient cells can result in epigenetic reprogramming or inducing or inhibiting cellular pathways in recipient cells.
Role of Melanoma-Derived EVs in Angiogenesis
Despite significant advances in EV research, knowledge of their role in melanoma-related angiogenesis is still very limited. Nevertheless, as mentioned above, the presence of various pro-and/or-antiangiogenic factors in melanoma-derived EVs has already been confirmed [5,6]. Several different mechanisms by which EVs may promote angiogenesis have also been investigated. In the following section, we review the available research data on the effect of melanoma-derived EVs on tumor angiogenesis and discuss their therapeutic and prognostic potential.
Transfer of proangiogenic factors and regulation of their expression in endothelial cells
Melanoma-derived EVs regulate angiogenesis itself or modulate related processes such as ECM remodeling. First, the urokinase-type plasminogen activator receptor (uPAR) carried by them increased the expression of EGFR, uPAR, and VE-cadherin, and activated the ERK1/2 pathway in recipient ECs [101]. Functional EGFR can also be directly transferred from melanoma to ECs via larger EVs, i.e., ectosomes (microvesicles), and such transfer depends on the phosphatidyl serine (PS) presence on the EV surface [101]. As a result, EGFR activates MAPK/Akt pathways in recipient cells and increases the expression of VEGF and its receptor VEGFR-2, thereby activating autocrine VEGF-VEGFR-2 signaling.
Moreover, overexpression of Wnt Family Member 5A (WNT5A) in several melanoma cell lines was associated with increased release of exosomes enriched in VEGF, IL-6, IL-8, and MMP-2 [102]. In addition, exosomes derived from melanoma cells overexpressing WNTA5A enhanced tube formation by ECs on Matrigel [102]. To follow up on matrix-degrading enzymes, membrane-type 1 matrix metalloproteinase (MT1-MMP) was also identified in melanoma-derived EVs [103].
Another proangiogenic factor identified in melanoma-derived EVs is tissue factor (TF). Alongside its procoagulant activity, TF can also stimulate signaling via the PAR-2 receptor, leading to upregulation of VEGF and enhanced angiogenesis. In addition, melanoma-derived EVs have been shown to contain more TF than melanocyte-derived EVs [104].
Finally, Hood et al. described the most complex 3D in vitro model of endothelial tissue to better mimic the microenvironment and morphology of ECs [105]. It was found that melanoma-derived exosomes are transferred between ECs via tunneling nanotubes. Moreover, melanoma-derived exosomes stimulated the formation of sprouting endothelial spheroids and the secretion of various proangiogenic cytokines. Interestingly, the secretion of IL-1α, FGF, GCS-F, TNFα, leptin, TGF-α, and VEGF, by endothelial spheroids correlated positively with the exosome dose used for incubation. These data provided evidence supporting the involvement of exosomes in endothelial angiogenic responses.
Induction of proangiogenic switch in bone marrow progenitor cells
Bone marrow-derived progenitor cells (BMPCs) are known to promote both, neoangiogenesis and remodeling of existing blood vessels. Peinado et al. showed that melanoma-derived exosomes induce a proangiogenic phenotype in BMPCs associated with the horizontal transfer of c-Kit, Tie-2, and Met oncoprotein [106]. Moreover, in melanoma-bearing B16F10 mice, the same exosomes enhanced the formation of pulmonary pre-metastatic niches with typical leaky vasculature that supports subsequent metastasis. Such an effect was not observed when exosomes with lower Met content were used, indicating that exosomal Met is a key factor determinant of the proangiogenic switch in BMPCs in melanoma. Similar findings were later made in another study in which the same B16F10 mice were also inoculated with Met-enriched exosomes [107]. It led to increased numbers of lung and femur metastases, while exosomes with lower Met expression did not induce significant changes.
Generation of cancer-associated fibroblasts (CAFs) displaying a proangiogenic phenotype
Melanoma-derived EVs display the potential to induce the differentiation of fibroblasts and/or ECs into CAFs. CAFs present in the TME can contribute to angiogenesis by directly altering the protein composition of TME but also by secretion matrix-degrading enzymes, for example. First of all, the transfer of lncRNA Gm26809 via melanoma-derived exosomes into normal fibroblasts induced their differentiation into CAFs [108]. Melanoma-derived exosomes carrying miR-155 have also been shown to induce CAF differentiation and downregulate the expression of the suppressor of cytokine signaling 1 (SOCS1) gene in recipient fibroblasts [109]. Downregulation of SOCS1 activated the Janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) signaling pathway, resulting in proangiogenic switch and increased expression of VEGF, FGF2, and MMP-9 in recipient CAFs. Moreover, exosomes from miR-155-overexpressing B16 mouse melanoma cells significantly alleviated tube formation in 2D assay by MS-1 endothelial cell line and increased microvessel density in mice melanoma xenografts. In contrast, the downregulation of exosomal miR-155 brought opposite results, both in vitro and in vivo.
Also, Yeon et al. showed in a microfluidic 3D microvascular model that exosomes from melanoma cells stimulate the differentiation of ECs into CAFs [110]. This process was associated with increased expression of endothelial to mesenchymal transition-related genes, i.e., alpha smooth muscle actin (α-SMA), fibroblast-specific protein-1 (FSP-1), MMP-9, N-/VE-cadherins, vimentin, and TGF-β.
Finally, the most recent study analyzed whether EVs derived from melanoma cells cultured under hypoxia affect the proangiogenic properties of CAFs [111]. Hypoxia led to the enrichment of melanoma-derived EVs with HSP90/phosphorylated inhibitor of NF-κB kinase (IKK) (p-IKKα/β) complex. Subsequent EV-mediated transfer of HSP90/p-IKKα/β to CAFs activated the IKK/IκB/NF-κB signaling pathway and promoted CXCL1 expression and secretion in CAFs. In the same study, conditioned media from CAFs that underwent the described proangiogenic switch were added to HUVECs and increased their proliferation and tube formation in the 2D Matrigel assay. Moreover, in a xenograft murine model, mice inoculated with hypoxic EVs showed larger tumors than those inoculated with normoxic EVs. Moreover, blocking of EV-associated HSP90 with tanespimycin significantly reduced the size of xenografts. This proved the involvement of CAFs and HSP90/IKK/NF-κB/CXCL1 axis in the regulation of melanoma angiogenesis by EVs.
Induction of proangiogenic properties of tumor-associated macrophages (TAMs)
The function of TAMs may also be regulated by melanoma-derived EVs. Due to the occurrence of their M1/M2 polarization, TAMs show varied functions in melanoma. Regarding angiogenesis, increased M2 polarization stimulates the formation of tumor vasculature. On the other hand, M1 TAMs have been shown to support the normalization of irregular vascular networks, which helps, for example, in the delivery of chemotherapeutics to tumor cells [112,113].
EVs are one of the mediators between melanoma cells and TAMs that may increase the latter’s proangiogenic properties. Back in 2016, Hood hypothesized that exosomes from melanoma cells increase the expression of granulocyte-macrophage colony-stimulating factor (GM-CSF) in ECs, and secreted GM-CSF increases the activity of HIF-2α in M2 TAMs [114]. HIF-2α further induces VEGFR-1 production, and activation of signaling pathways mediated by VEGF. Unfortunately, this hypothesis has not been confirmed experimentally. However, in a later study, Jarosz-Biej et al. [115] showed on melanoma tumor tissues that higher blood vessel density correlates positively with increased numbers of M1 rather than M2 TAMs.
Recently, Parikh et al. investigated the properties of melanosomes, a specific, melanin-transporting population of EVs derived from MNT-1 cells [116]. Unlike other EVs that are utilized by the recipient cell, melanosomes remain intact and can be further transferred to another cell. The isolated MNT1-derived melanosomes were used to treat CAFs and were then re-isolated and used to treat macrophages. From the very beginning, they carried AKT1, which stimulated mTOR-dependent VEGF secretion by macrophages, promoting angiogenesis in the murine model. Furthermore, the same study used human melanoma specimens and positively correlated macrophages histologically co-localized with AKT1 with more progressive disease. Moreover, samples from patients unresponsive to immunotherapy were enriched in macrophages expressing melanosome markers.
Role of melanoma-derived EVs in lymphangiogenesis
Besides classical angiogenesis, melanoma-derived EVs regulate lymphatic vessel formation, as first shown in premetastatic niches in mice [117]. The observed effect was attributed to the nerve growth factor receptor (NGFR) transferred via EVs and activated ERK and NF-κB signaling in recipient lymphatic ECs. Consistently, EVs not expressing NGFR reduced the number of lymph node metastases and improved the survival of B16 mice. Moreover, lymphatic ECs were shown to overexpress intracellular adhesion molecules (ICAM-1) after EV treatment. Overexpression of ICAM-1 contributed to increased lymphangiogenesis and adhesion of melanoma cells.
Another study also found interactions between melanoma-derived EVs and lymphatic EC, this time mediated by vascular cell adhesion molecule 1 (VCAM-1) [118]. EVs increased lymphatic ECs proliferation and lymph node remodeling. Moreover, EVs transferred various melanoma antigens and MHC-1 molecules, and their subsequent presentation by lymphatic ECs led to cytotoxic T cells apoptotic death and subsequent inhibition of the immune response. The role of melanoma-derived EVs in angiogenesis is summarized in Fig. 4.
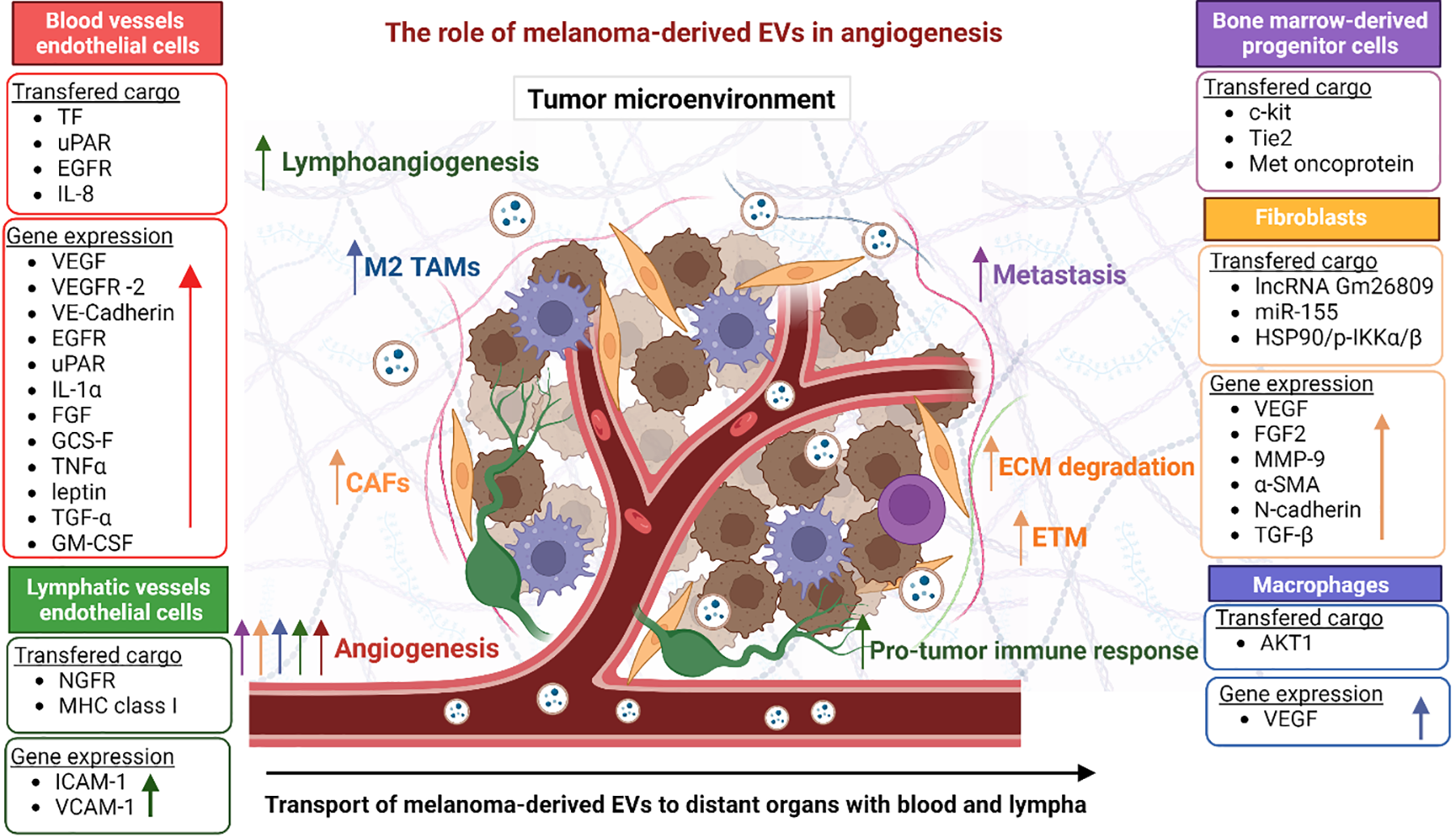
Figure 4: Summary of the role of melanoma-derived EVs in angiogenesis by affecting cells present in the TME. Cell types and their function are color-coded. The melanoma microenvironment consists of several types of cells, including immune system cells (TAMs, neutrophils, T cells) and ECs. These cells cooperate in multiple processes involved in melanoma progression, one of which is angiogenesis. The coexistence of cells forms paracrine loops regulating ECM remodeling, lymphoangiogenesis, and importantly the formation of new blood vessels and their maturation. The figure was prepared under the license in BioRender: Scientific Image and Illustration Software.
Clinical Potential of EVs in Antiangiogenic Therapies
Angiogenesis is undoubtedly a key step in melanoma progression, enabling faster tumor growth and local/distant metastases. Unfortunately, many diagnoses are made after the disease has reached this point, hindering the effectiveness of common therapeutic approaches. For these reasons, the majority of melanoma treatment strategies involve antiangiogenic drugs that target VEGF, VEGFR, FGFR, PDGFR, or integrins (summary in Table 1).
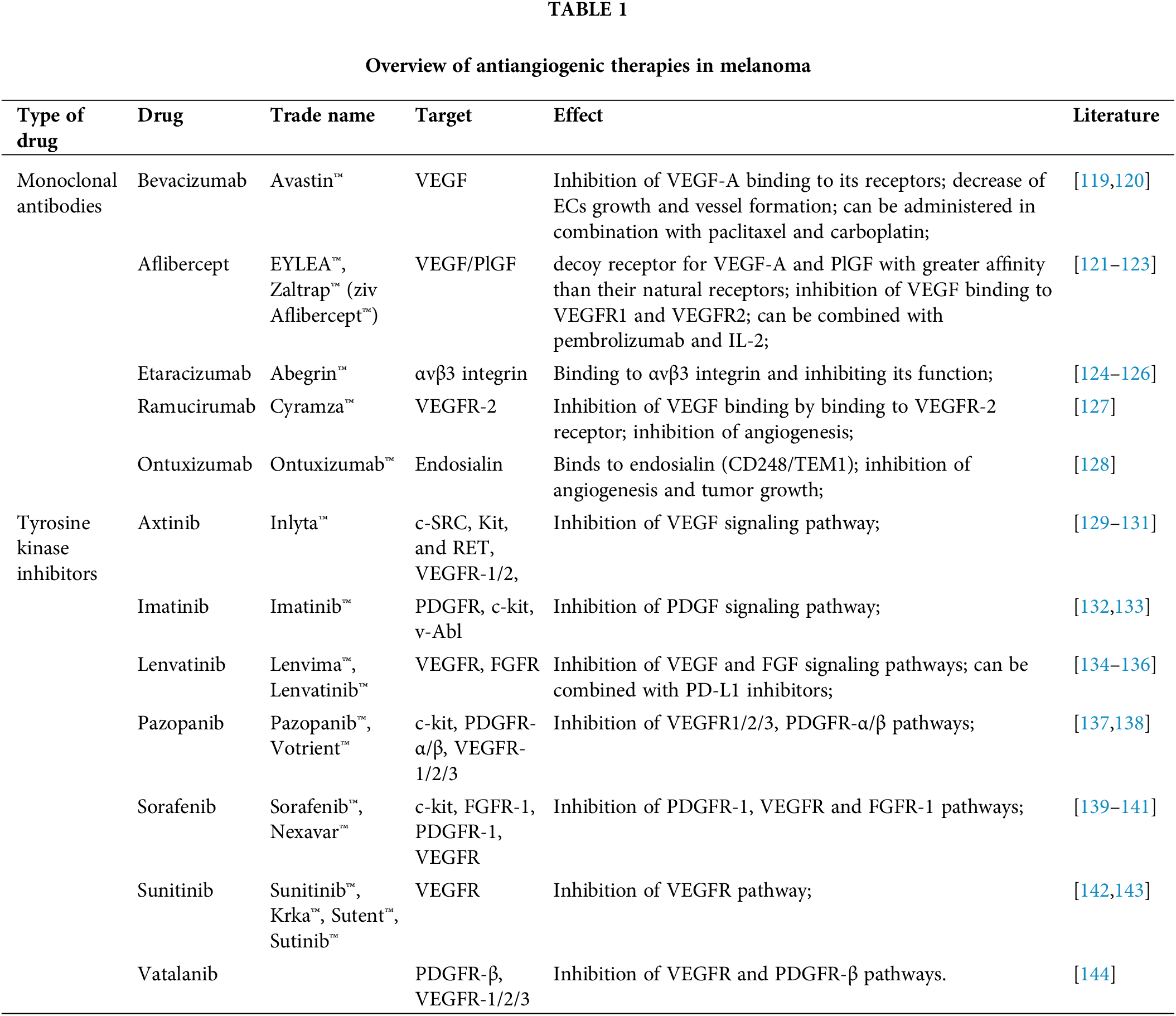
Although a modest improvement in overall survival has been observed following the administration of antiangiogenic therapies, there are still unresponsive patients who have developed resistance over time. EVs likely contribute to the failure of antiangiogenic therapies. In non-melanoma cancers, such as breast, colorectal, and renal cancers or glioblastoma, EVs have been shown to transfer different variants of VEGF with a much lower affinity for antiangiogenic drugs (mainly bevacizumab) than soluble VEGF in plasma. On the other hand, VEGF variants transferred via EVs show higher affinity for VEGFRs on ECs, which may represent a detour used by tumor cells to sustain angiogenesis [145].
However, there is another side to the EV story. The specific properties of EVs can be used to improve the effectiveness of antiangiogenic therapies in melanoma. First of all, abrogation of proangiogenic EV release in combination with antiangiogenic drugs could restore sensitivity to, for example, drugs targeting the VEGF/VEGFR axis. Known inhibitors of EV release/uptake by recipient cells are shown in Table 2 and have been thoroughly reviewed [146]. Unfortunately, no studies or clinical trials have been conducted on the combined use of EV inhibitors and anti-angiogenic drugs. However, recently, EVs in the plasma of healthy individuals were shown to transfer mostly anti-angiogenic proteins which suppress the angiogenic properties of recipient ECs [147]. On the other hand, EVs from patients with head and neck cancer increased migration and proliferation of ECs and their tube-forming potential. This suggests that targeting EV release may be an indirect way to abrogate tumor angiogenesis.
Another thing to consider is the development of new drugs (antibodies, etc.) binding specifically to the different variants of proangiogenic factors expressed in EVs. Importantly, not only tumor-derived EVs shall be evaluated as potential targets for antiangiogenic therapies in melanoma. The function of other TEM cells such as ECs, CAFs, or macrophages, strongly depends on intercellular communication mediated by EVs. EVs, which, for example, induce polarization of M1 macrophages, should therefore be a focus of further research on tumor angiogenesis.
Moreover, EVs are biocompatible, non-toxic, and have high bioavailability. Because of these properties, they are used for drug delivery after being loaded with various drugs, including antiangiogenic agents used in melanoma treatment (Table 1). However, although EVs do not exhibit immunotoxicity, they can exhibit varying degrees of immunogenicity, which can potentially contribute to their increased clearance. The immunogenicity of EVs may be due to their surface molecular composition, as well as their internal cargo, cellular origin, dosage and infusion rate, etc. To mitigate this phenomenon, it is possible to use less differentiated cells as a source of EVs, modification EV cargo (removal of immune response-inducing molecules such as MHC-I or introduction of immune suppressors–complement regulator, PEG), reduction of dose and infusion rates, or use of smaller particles for drug delivery [148].
As mentioned above, no clinical trials strictly utilizing EVs in antiangiogenic therapy have been registered to date. However, the potential of various EV-based therapeutic strategies has been proven in several types of cancers and other diseases Almost 400 diagnostic or therapeutic trials utilizing EVs are registered on clinicaltrials.gov, of which more than 60 use EV therapy as the primary intervention, and articles have been already published extensively discussing this issue [149,150]. Currently, most trials focus on lung diseases, mainly due to the COVID-19 pandemic, although EVs are also used to treat acute respiratory distress syndrome and non-COVID-19 infections. Other important therapeutic applications of EVs include, among others, anti-rejection therapy post organ transplantation, gastroenterological diseases (inflammatory bowel disease, including Crohn’s and ulcerative colitis), hypercholesterolemia or nervous system conditions (Alzheimer’s, depression, neuralgia, or stroke) [149,150].
Finally, regenerative medicine is a field where EVs also show well-documented therapeutic potential. They are used for wound healing and regeneration (including burns, venous trophic lesions, etc.), treatments of bone defects and meniscal injuries, as well as muscle regeneration after myocardial infarction [149,150]. Importantly, tissue regeneration most often includes angiogenesis, so such trials indirectly indicate the potential of EV-based therapy in various conditions involving angiogenesis. For instance, there is a clinical trial using autologous plasma-derived exosomes for the management of most severe cutaneous ulcers (NCT02565264).
In cancer, however, enhanced angiogenesis is one of the factors contributing to cancer progression. The potential of EVs in antiangiogenic therapy has not yet been evaluated in clinical settings. Nevertheless, since the early 2000s, EV-oriented clinical trials have been developed for various cancers. The first trials used EVs as a vaccine to boost the anti-tumor immune response in colon cancer [151], non-small cell lung cancer [152] and melanoma [153]. Although they involved a small number of patients and showed unsatisfactory results, they clearly indicated the possibility and safety of therapeutic EV administration. In terms of ongoing trials, EV are currently being tested for the treatment of advanced hepatocellular carcinoma and liver metastasis of gastric and colorectal cancer (NCT05375604, EVs loaded with STAT6 antisense oligonucleotides), metastatic pancreatic cancer with a KRAS G12D mutation (NCT03608631, EVs loaded with KRAS G12D siRNA), and colon cancer (EVs loaded with curcumin).
None of the aforementioned trials was focused on angiogenesis, leaving the area unexplored for now. However, the data from basic research suggests that the antiangiogenic application of EVs may be a way to control or constrain the impact of tumor angiogenesis during cancer progression.
Conclusions and Future Perspectives
Despite ongoing research in the EV field, there is a shortage of studies on melanoma-derived EVs in terms of their role and potential therapeutic application in antiangiogenic therapies. In particular, in vivo and clinical studies are lacking. Although the pro-/antiangiogenic action of EVs has been demonstrated in simpler models, there are still some limitations preventing the wider use of EVs in antiangiogenic therapies. For example, isolated/drug-loaded EVs must be strictly standardized in terms of their cellular source, concentration, purity, stability, etc. That should be followed by of the application of the most appropriate and efficient protocol for drug loading (Fig. 5). It is also necessary to ensure accurate biodistribution within the tumor vasculature, which involves providing therapeutic EVs with targeting molecules and assessing various routes of their administration. These issues make large-scale production of therapeutic EVs a major challenge. At that point, therapies based on artificial liposome-based EV-mimicking particles appear to be more within reach [174], as their production can be easily standardized and more widely commercialized. Another approach should include investigating which key proangiogenic components of EV cargo or signaling pathways activated by EVs in recipient cells during angiogenesis should be targeted by novel antiangiogenic therapies.
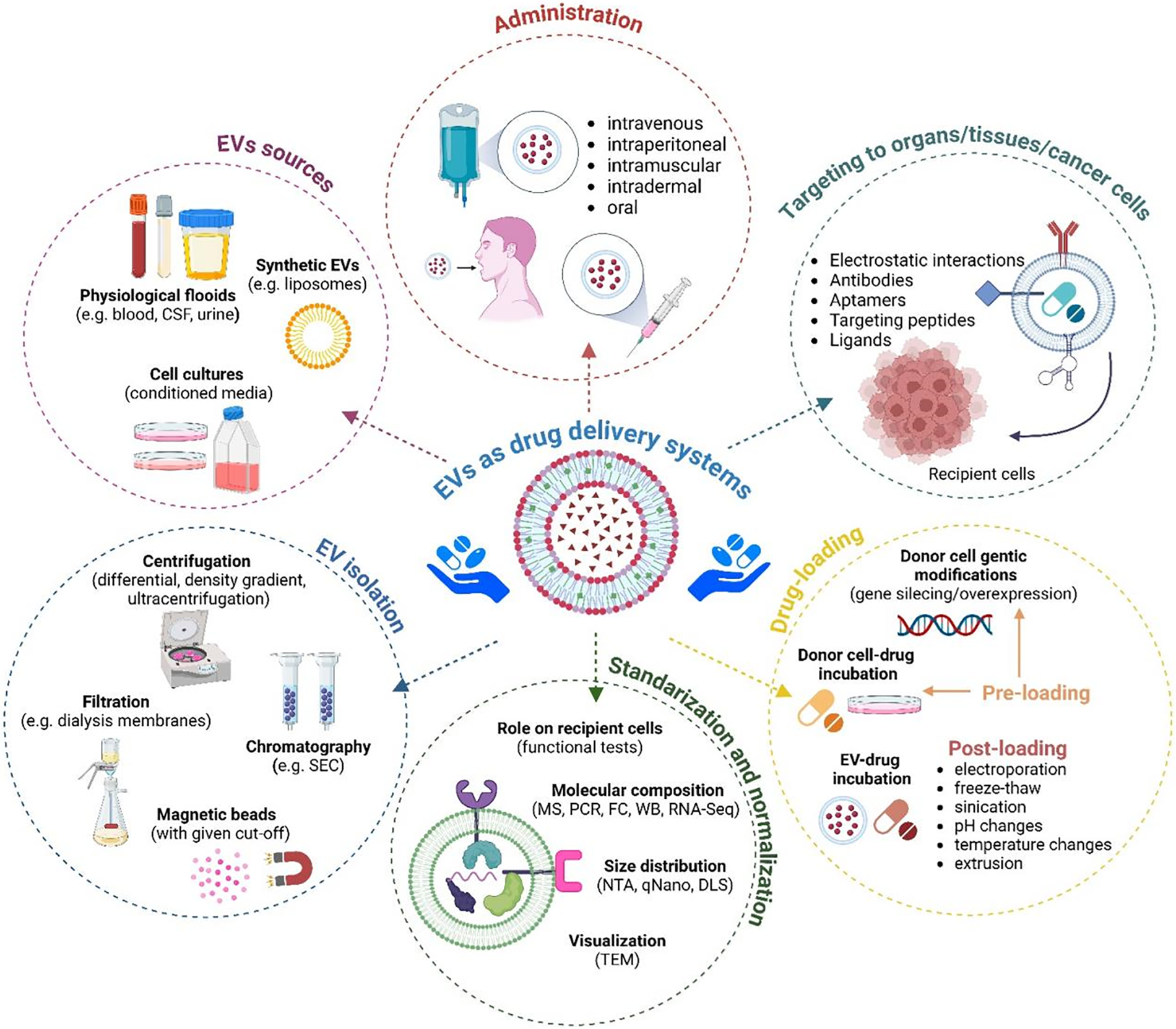
Figure 5: Various aspects of using EVs as drug delivery systems. The figure summarizes methods for isolating EVs from various sources, methods for standardizing and normalizing, methods for drug-loading and targeting them to recipient cells, and routes of administration. Used abbreviations: CSF–cerebrospinal fluid, DLS–dynamic light scattering, FC–flow cytometry, MS–mass spectrometry, NTA–nano tracking analysis, PCR–polymerase chain reaction, RNA seq–RNA sequencing, SEC–size exclusion chromatography, TEM–transmission electron microscopy, WB–western blotting. The figure was prepared under the license in BioRender: Scientific Image and Illustration Software.
Acknowledgement: All figures presented in the manuscript were prepared under the license in BioRender: Scientific Image and Illustration Software.
Funding Statement: The writing of this manuscript was supported by grants from the Jagiellonian University, Poland (N18/DBS/000007) and the Polish National Science Centre (2018/31/N/NZ4/03787).
Author Contributions: The authors confirm contribution to the paper as follows: draft manuscript preparation: Magdalena Wilczak, Magdalena Surman; figure preparation: Magdalena Wilczak; revision and editing of manuscript: Małgorzata Przybyło. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. Saman H, Raza SS, Uddin S, Rasul K. Inducing angiogenesis, a key step in cancer vascularization, and treatment approaches. Cancers. 2020;12(5):1172. doi:10.3390/cancers12051172. [Google Scholar] [PubMed] [CrossRef]
2. Marech I, Leporini C, Ammendola M, Porcelli M, Gadaleta CD, Russo E, et al. Classical and non-classical proangiogenic factors as a target of antiangiogenic therapy in tumor microenvironment. Cancer Lett. 2016;380(1):216–26. doi:10.1016/j.canlet.2015.07.028. [Google Scholar] [PubMed] [CrossRef]
3. Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci. 2020;77(9):1745–70. doi:10.1007/s00018-019-03351-7. [Google Scholar] [PubMed] [CrossRef]
4. Liu ZL, Chen HH, Zheng LL, Sun LP, Shi L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Sig Transduct Target Ther. 2023;8:198. doi:10.1038/s41392-023-01460-1. [Google Scholar] [PubMed] [CrossRef]
5. Ye ZW, Yu ZL, Chen G, Jia J. Extracellular vesicles in tumor angiogenesis and resistance to anti-angiogenic therapy. Cancer Sci. 2023;114(7):2739–49. doi:10.1111/cas.15801. [Google Scholar] [PubMed] [CrossRef]
6. Ateeq M, Broadwin M, Sellke FW, Abid MR. Extracellular vesicles’ role in angiogenesis and altering angiogenic signaling. Med Sci. 2024;12(1):4. doi:10.3390/medsci12010004. [Google Scholar] [PubMed] [CrossRef]
7. Lopes J, Rodrigues CMP, Gaspar MM, Reis CP. Melanoma management: from epidemiology to treatment and latest advances. Cancers. 2022;14(19):4652. doi:10.3390/cancers14194652. [Google Scholar] [PubMed] [CrossRef]
8. Arnold M, Singh D, Laversanne M, Vignat J, Vaccarella S, Meheus F, et al. Global burden of cutaneous melanoma in 2020 and projections to 2040. JAMA Dermatol. 2022;158(5):495–503. doi:10.1001/jamadermatol.2022.0160. [Google Scholar] [PubMed] [CrossRef]
9. Waseh S, Lee JB. Advances in melanoma: epidemiology, diagnosis, and prognosis. Front Med. 2023;10:1268479. doi:10.3389/fmed.2023.1268479. [Google Scholar] [PubMed] [CrossRef]
10. Kaštelan S, Mrazovac ZD, Ivanković M, Marković I, Gverović AA. Liver metastasis in uveal melanoma-treatment options and clinical outcome. Front Biosci (Landmark Ed). 2022;27(2):72. doi:10.31083/j.fbl2702072. [Google Scholar] [PubMed] [CrossRef]
11. Chen Z, Han F, Du Y, Shi H, Zhou W. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther. 2023;8(1):70. doi:10.1038/s41392-023-01332-8. [Google Scholar] [PubMed] [CrossRef]
12. Kling L, Schreiber A, Eckardt KU, Kettritz R. Hypoxia-inducible factors not only regulate but also are myeloid-cell treatment targets. J Leukoc Biol. 2021;110(1):61–75. doi:10.1002/JLB.4RI0820-535R. [Google Scholar] [PubMed] [CrossRef]
13. Lee SB, Ko A, Oh YT, Shi P, D’Angelo F, Frangaj B, et al. Proline hydroxylation primes protein kinases for autophosphorylation and activation. Mol Cell. 2020;79(3):376–89.e8. doi:10.1016/j.molcel.2020.06.021. [Google Scholar] [PubMed] [CrossRef]
14. Volkova YL, Pickel C, Jucht AE, Wenger RH, Scholz CC. The asparagine hydroxylase FIH: a unique oxygen sensor. Antioxid Redox Signal. 2022;37(13–15):913–35. doi:10.1089/ars.2022.0003. [Google Scholar] [PubMed] [CrossRef]
15. Fan S, Wang J, Yu G, Rong F, Zhang D, Xu C, et al. TET is targeted for proteasomal degradation by the PHD-pVHL pathway to reduce DNA hydroxymethylation. J Biol Chem. 2020;295(48):16299–313. doi:10.1074/jbc.RA120.014538. [Google Scholar] [PubMed] [CrossRef]
16. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia. 2015;3:83–92. doi:10.2147/HP.S93413. [Google Scholar] [PubMed] [CrossRef]
17. Tafani M, Pucci B, Russo A, Schito L, Pellegrini L, Perrone GA, et al. Modulators of HIF1α and NFKB in cancer treatment: is it a rational approach for controlling malignant progression? Front Pharmacol. 2013;4:13. doi:10.3389/fphar.2013.00013. [Google Scholar] [PubMed] [CrossRef]
18. Eelen G, Treps L, Li X, Carmeliet P. Basic and therapeutic aspects of angiogenesis updated. Circ Res. 2020;127(2):310–29. doi:10.1161/CIRCRESAHA.120.316851. [Google Scholar] [PubMed] [CrossRef]
19. Dudley AC, Griffioen AW. Pathological angiogenesis: mechanisms and therapeutic strategies. Angiogenesis. 2023;26(3):313–47. doi:10.1007/s10456-023-09876-7. [Google Scholar] [PubMed] [CrossRef]
20. Naito H, Iba T, Takakura N. Mechanisms of new blood-vessel formation and proliferative heterogeneity of endothelial cells. Int Immunol. 2020;32(5):295–305. doi:10.1093/intimm/dxaa008. [Google Scholar] [PubMed] [CrossRef]
21. Ahmad A, Nawaz MI. Molecular mechanism of VEGF and its role in pathological angiogenesis. J Cell Biochem. 2022 Dec;123(12):1938–65. doi:10.1002/jcb.30344. [Google Scholar] [PubMed] [CrossRef]
22. Ghalehbandi S, Yuzugulen J, Pranjol MZI, Pourgholami MH. The role of VEGF in cancer-induced angiogenesis and research progress of drugs targeting VEGF. Eur J Pharmacol. 2023;949:175586. doi:10.1016/j.ejphar.2023.175586. [Google Scholar] [PubMed] [CrossRef]
23. Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103:159–65. doi:10.1172/JCI5028. [Google Scholar] [PubMed] [CrossRef]
24. Gorski DH, Leal AD, Goydos JS. Differential expression of vascular endothelial growth factor—a isoforms at different stages of melanoma progression. J Am Coll Surg. 2003;197:408–18. doi:10.1016/S1072-7515(03)00388-0. [Google Scholar] [PubMed] [CrossRef]
25. Ribatti D, Nico B, Floris C, Mangieri D, Piras F, Ennas MG, et al. Microvascular density, vascular endothelial growth factor immunoreactivity in tumor cells, vessel diameter and intussusceptive microvascular growth in primary melanoma. Oncol Rep. 2005;14(1):81–4. [Google Scholar] [PubMed]
26. Rofstad EK, Danielsen T. Hypoxia-induced angiogenesis and vascular endothelial growth factor secretion in human melanoma. Br J Cancer. 1998;77(6):897–902. doi:10.1038/bjc.1998.148. [Google Scholar] [PubMed] [CrossRef]
27. Rajabi P, Neshat A, Mokhtari M, Rajabi MA, Eftekhari M, Tavakoli P. The role of VEGF in melanoma progression. J Res Med Sci. 2012;17(6):534–9. [Google Scholar] [PubMed]
28. Hirakawa S, Kodama S, Kunstfeld R, Kajiya K, Brown LF, Detmar M. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med. 2005;201(7):1089–99. doi:10.1084/jem.20041896. [Google Scholar] [PubMed] [CrossRef]
29. Kwon S, Velasquez FC, Sevick-Muraca EM. Near-infrared fluorescence lymphatic imaging in vascular endothelial growth factor-C overexpressing murine melanoma. Biomed Opt Express. 2018;9(10):4631–7. doi:10.1364/BOE.9.004631. [Google Scholar] [PubMed] [CrossRef]
30. Špirić Z, Eri Ž, Erić M. Lymphatic vessel density and VEGF-C expression as independent predictors of melanoma metastases. J Plast Reconstr Aesthet Surg. 2017;70(11):1653–9. doi:10.1016/j.bjps.2017.06.040. [Google Scholar] [PubMed] [CrossRef]
31. Špirić Z, Eri Ž, Erić M. Significance of Vascular Endothelial Growth Factor (VEGF)-C and VEGF-D in the progression of Cutaneous Melanoma. Int J Surg Pathol. 2015;23(8):629–37. doi:10.1177/1066896915583694. [Google Scholar] [PubMed] [CrossRef]
32. Lund AW, Duraes FV, Hirosue S, Raghavan VR, Nembrini C, Thomas SN, et al. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep. 2012;1(3):191–9. doi:10.1016/j.celrep.2012.01.005. [Google Scholar] [PubMed] [CrossRef]
33. Lee C, Chen R, Sun G, Liu X, Lin X, He C, et al. VEGF-B prevents excessive angiogenesis by inhibiting FGF2/FGFR1 pathway. Signal Transduct Target Ther. 2023;8(1):305. doi:10.1038/s41392-023-01539-9. [Google Scholar] [PubMed] [CrossRef]
34. Guérit E, Arts F, Dachy G, Boulouadnine B, Demoulin JB. PDGF receptor mutations in human diseases. Cell Mol Life Sci. 2021;78(8):3867–81. doi:10.1007/s00018-020-03753-y. [Google Scholar] [PubMed] [CrossRef]
35. Barnhill RL, Xiao M, Graves D, Antoniades HN. Expression of platelet-derived growth factor (PDGF)-A, PDGF-B and the PDGF-alpha receptor, but not the PDGF-beta receptor, in human malignant melanoma in vivo. Br J Dermatol. 1996;135(6):898–904. doi:10.1046/j.1365-2133.1996.d01-1092.x. [Google Scholar] [PubMed] [CrossRef]
36. Ruffini F, Levati L, Graziani G, Caporali S, Atzori MG, D’Atri S, et al. Platelet-derived growth factor-C promotes human melanoma aggressiveness through activation of neuropilin-1. Oncotarget. 2017;8(40):66833–48. doi:10.18632/oncotarget.18706. [Google Scholar] [PubMed] [CrossRef]
37. Robinson SP, Ludwig C, Paulsson J, Ostman A. The effects of tumor-derived platelet-derived growth factor on vascular morphology and function in vivo revealed by susceptibility MRI. Int J Cancer. 2008;122(7):1548–56. doi:10.1002/ijc.23279. [Google Scholar] [PubMed] [CrossRef]
38. Faraone D, Aguzzi MS, Toietta G, Facchiano AM, Facchiano F, Magenta A, et al. Platelet-derived growth factor-receptor alpha strongly inhibits melanoma growth in vitro and in vivo. Neoplasia. 2009;11:732–42. doi:10.1593/neo.09408. [Google Scholar] [PubMed] [CrossRef]
39. Forsberg K, Valyi-Nagy I, Heldin CH, Herlyn M, Westermark B. Platelet-derived growth factor (PDGF) in oncogenesis: development of a vascular connective tissue stroma in xenotransplanted human melanoma producing PDGF-BB. Proc Natl Acad Sci U S A. 1993;90(2):393–7. doi:10.1073/pnas.90.2.393. [Google Scholar] [PubMed] [CrossRef]
40. Filimon A, Preda IA, Boloca AF, Negroiu G. Interleukin-8 in melanoma pathogenesis, prognosis and therapy—an integrated view into other neoplasms and chemokine networks. Cells. 2021;11(1):120. doi:10.3390/cells11010120. [Google Scholar] [PubMed] [CrossRef]
41. Payne AS, Cornelius LA. The role of chemokines in melanoma tumor growth and metastasis. J Invest Dermatol. 2002;118(6):915–22. doi:10.1046/j.1523-1747.2002.01725.x. [Google Scholar] [PubMed] [CrossRef]
42. Singh RK, Varney ML, Bucana CD, Johansson SL. Expression of interleukin-8 in primary and metastatic malignant melanoma of the skin. Melanoma Res. 1999;9(4):383–7. doi:10.1097/00008390-199908000-00007. [Google Scholar] [PubMed] [CrossRef]
43. Dömer D, Walther T, Möller S, Behnen M, Laskay T. Neutrophil extracellular traps activate proinflammatory functions of human neutrophils. Front Immunol. 2021;12:636954. doi:10.3389/fimmu.2021.636954. [Google Scholar] [PubMed] [CrossRef]
44. Matsushima K, Yang D, Oppenheim JJ. Interleukin-8: an evolving chemokine. Cytokine. 2022;153:155828. doi:10.1016/j.cyto.2022.155828. [Google Scholar] [PubMed] [CrossRef]
45. Li A, Varney ML, Valasek J, Godfrey M, Dave BJ, Singh RK. Autocrine role of interleukin-8 in induction of endothelial cell proliferation, survival, migration and MMP-2 production and angiogenesis. Angiogenesis. 2005;8(1):63–71. doi:10.1007/s10456-005-5208-4. [Google Scholar] [PubMed] [CrossRef]
46. Shoshan E, Braeuer RR, Kamiya T, Mobley AK, Huang L, Vasquez ME, et al. NFAT1 directly regulates IL8 and MMP3 to promote melanoma tumor growth and metastasis. Cancer Res. 2016;76(11):3145–55. doi:10.1158/0008-5472.CAN-15-2511. [Google Scholar] [PubMed] [CrossRef]
47. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. 2020;20:485–503. doi:10.1038/s41568-020-0281-y. [Google Scholar] [PubMed] [CrossRef]
48. Schedel F, Mayer-Hain S, Pappelbaum KI, Metze D, Stock M, Goerge T, et al. Evidence and impact of neutrophil extracellular traps in malignant melanoma. Pigment Cell Melanoma Res. 2020;33(1):63–73. doi:10.1111/pcmr.12818. [Google Scholar] [PubMed] [CrossRef]
49. Asokan S, Bandapalli OR. CXCL8 signaling in the tumor microenvironment. Adv Exp Med Biol. 2021;1302:25–39. doi:10.1007/978-3-030-62658-7. [Google Scholar] [CrossRef]
50. Ardizzone A, Bova V, Casili G, Repici A, Lanza M, Giuffrida R, et al. Role of basic fibroblast growth factor in cancer: biological activity, targeted therapies, and prognostic value. Cells. 2023;12(7):1002. doi:10.3390/cells12071002. [Google Scholar] [PubMed] [CrossRef]
51. Li C, Kuang K, Du J, Eymin B, Jia T. Far beyond anti-angiogenesis: benefits for anti-basicFGF therapy in cancer. Biochim Biophys Acta Mol Cell Res. 2022;1869(7):119253. doi:10.1016/j.bbamcr.2022.119253. [Google Scholar] [PubMed] [CrossRef]
52. Wu Z, Bian Y, Chu T, Wang Y, Man S, Song Y, et al. The role of angiogenesis in melanoma: clinical treatments and future expectations. Front Pharmacol. 2022;13:1028647. doi:10.3389/fphar.2022.1028647. [Google Scholar] [PubMed] [CrossRef]
53. Hosaka K, Yang Y, Nakamura M, Andersson P, Yang X, Zhang Y, et al. Dual roles of endothelial FGF-2-FGFR1-PDGF-BB and perivascular FGF-2-FGFR2-PDGFRβ signaling pathways in tumor vascular remodeling. Cell Discov. 2018;4:3. doi:10.1038/s41421-017-0002-1. [Google Scholar] [PubMed] [CrossRef]
54. Reiland J, Kempf D, Roy M, Denkins Y, Marchetti D. FGF2 binding, signaling, and angiogenesis are modulated by heparanase in metastatic melanoma cells. Neoplasia. 2006;8(7):596–606. doi:10.1593/neo.06244. [Google Scholar] [PubMed] [CrossRef]
55. Czyz M. Fibroblast growth factor receptor signaling in skin cancers. Cells. 2019;8(6):540. doi:10.3390/cells8060540. [Google Scholar] [PubMed] [CrossRef]
56. Streit S, Mestel DS, Schmidt M, Ullrich A, Berking C. FGFR4 Arg388 allele correlates with tumour thickness and FGFR4 protein expression with survival of melanoma patients. Br J Cancer. 2006;94(12):1879–86. doi:10.1038/sj.bjc.6603181. [Google Scholar] [PubMed] [CrossRef]
57. Parmar D, Apte M. Angiopoietin inhibitors: a review on targeting tumor angiogenesis. Eur J Pharmacol. 2021;899:174021. doi:10.1016/j.ejphar.2021.174021. [Google Scholar] [PubMed] [CrossRef]
58. Nasarre P, Thomas M, Kruse K, Helfrich I, Wolter V, Deppermann C, et al. Host-derived angiopoietin-2 affects early stages of tumor development and vessel maturation but is dispensable for later stages of tumor growth. Cancer Res. 2009;69(4):1324–33. doi:10.1158/0008-5472.CAN-08-3030. [Google Scholar] [PubMed] [CrossRef]
59. Leong A, Kim M. The Angiopoietin-2 and TIE pathway as a therapeutic target for enhancing antiangiogenic therapy and immunotherapy in patients with advanced cancer. Int J Mol Sci. 2020;21(22):8689. doi:10.3390/ijms21228689. [Google Scholar] [PubMed] [CrossRef]
60. Cazzato G, Ingravallo G, Ribatti D. Angiogenesis still plays a crucial role in human melanoma progression. Cancers. 2024;16(10):1794. doi:10.3390/cancers16101794. [Google Scholar] [PubMed] [CrossRef]
61. Javelaud D, Alexaki VI, Mauviel A. Transforming growth factor-beta in cutaneous melanoma. Pigment Cell Melanoma Res. 2008;21(2):123–32. doi:10.1111/j.1755-148X.2008.00450.x. [Google Scholar] [PubMed] [CrossRef]
62. Van Belle P, Rodeck U, Nuamah I, Halpern AC, Elder DE. Melanoma-associated expression of transforming growth factor-beta isoforms. Am J Pathol. 1996;148(6):1887–94. [Google Scholar] [PubMed]
63. Liu D, Kumar S, Ashworth J, Ali K, Fadel A, Guo B, et al. CD105 (Endoglina potential anticancer therapeutic inhibits mitogenesis and Map kinase pathway activation. Anticancer Res. 2021;41(3):1219–29. doi:10.21873/anticanres.14879. [Google Scholar] [PubMed] [CrossRef]
64. Bodey B, Bodey BJr, Siegel SE, Kaiser HE. Immunocytochemical detection of endoglin is indicative of angiogenesis in malignant melanoma. Anticancer Res. 1998;18(4A):2701–10. [Google Scholar] [PubMed]
65. Rothhammer T, Bataille F, Spruss T, Eissner G, Bosserhoff AK. Functional implication of BMP4 expression on angiogenesis in malignant melanoma. Oncogene. 2007;26(28):4158–70. doi:10.1038/sj.onc.1210182. [Google Scholar] [PubMed] [CrossRef]
66. Rothhammer T, Poser I, Soncin F, Bataille F, Moser M, Bosserhoff AK. Bone morphogenic proteins are overexpressed in malignant melanoma and promote cell invasion and migration. Cancer Res. 2005;65(2):448–56. doi:10.1158/0008-5472.448.65.2. [Google Scholar] [CrossRef]
67. Odorisio T, Cianfarani F, Failla CM, Zambruno G. The placenta growth factor in skin angiogenesis. J Dermatol Sci. 2006;41(1):11–9. doi:10.1016/j.jdermsci.2005.08.008. [Google Scholar] [PubMed] [CrossRef]
68. Mahabeleshwar GH, Byzova TV. Angiogenesis in melanoma. Semin Oncol. 2007;34(6):555–65. doi:10.1053/j.seminoncol.2007.09.009. [Google Scholar] [PubMed] [CrossRef]
69. Lacal PM, Failla CM, Pagani E, Odorisio T, Schietroma C, Falcinelli S, et al. Human melanoma cells secrete and respond to placenta growth factor and vascular endothelial growth factor. J Invest Dermatol. 2000;115(6):1000–7. doi:10.1046/j.1523-1747.2000.00199.x. [Google Scholar] [PubMed] [CrossRef]
70. Luttun A, Autiero M, Tjwa M, Carmeliet P. Genetic dissection of tumor angiogenesis: are PIGF and VEGFR-1 novel anti-cancer targets? Biochim Biophys Acta. 2004;1654(1):79–94. doi:10.1016/j.bbcan.2003.09.002. [Google Scholar] [PubMed] [CrossRef]
71. Pagani E, Ruffini F, Antonini Cappellini GC, Scoppola A, Fortes C, Marchetti P, et al. Placenta growth factor and neuropilin-1 collaborate in promoting melanoma aggressiveness. Int J Oncol. 2016;48(4):1581–9. doi:10.3892/ijo.2016.3362. [Google Scholar] [PubMed] [CrossRef]
72. Bassiouni W, Ali MAM, Schulz R. Multifunctional intracellular matrix metalloproteinases: implications in disease. FEBS J. 2021;288(24):7162–82. doi:10.1111/febs.15701. [Google Scholar] [PubMed] [CrossRef]
73. de Almeida LGN, Thode H, Eslambolchi Y, Chopra S, Young D, Gill S, et al. Matrix Metalloproteinases: from molecular mechanisms to physiology, pathophysiology, and pharmacology. Pharmacol Rev. 2022;74(3):712–68. doi:10.1124/pharmrev.121.000349. [Google Scholar] [PubMed] [CrossRef]
74. Zeng Y, Gao M, Lin D, Du G, Cai Y. Prognostic and immunological roles of MMP-9 in pan-cancer. Biomed Res Int. 2022;2022:2592962. doi:10.1155/2022/2592962. [Google Scholar] [PubMed] [CrossRef]
75. Hofmann UB, Westphal JR, Van Muijen GN, Ruiter DJ. Matrix metalloproteinases in human melanoma. J Invest Dermatol. 2000;115(3):337–44. doi:10.1046/j.1523-1747.2000.00068.x. [Google Scholar] [PubMed] [CrossRef]
76. Hofmann UB, Westphal JR, Zendman AJ, Ruiter DJ, Van Muijen GN. Expression and activation of matrix metalloproteinase-2 (MMP-2) and its co-localization with membrane-type 1 matrix metalloproteinase (MT1-MMP) correlate with melanoma progression. J Pathol. 2000;191(3):245–56. doi:10.1046/j.1523-1747.2000.00068.x. [Google Scholar] [CrossRef]
77. Luca M, Huang S, Gershenwald JE, Singh RK, Reich R, Bar-Eli M. Expression of interleukin-8 by human melanoma cells up-regulates MMP-2 activity and increases tumor growth and metastasis. Am J Pathol. 1997;151:1105–13. [Google Scholar] [PubMed]
78. Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003;3(6):411–21. doi:10.1038/nrc1092. [Google Scholar] [PubMed] [CrossRef]
79. Yu Q, Stamenkovic I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999;13:35–48. doi:10.1101/gad.13.1.35. [Google Scholar] [PubMed] [CrossRef]
80. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumour microenvironment. Cell. 2010;141:52–67. doi:10.1016/j.cell.2010.03.015. [Google Scholar] [PubMed] [CrossRef]
81. Liang X, Sun R, Zhao X, Zhang Y, Gu Q, Dong X, et al. Rictor regulates the vasculogenic mimicry of melanoma via the AKT‐MMP‐2/9 pathway. J Cell Mol Med. 2017;21:3579–91. doi:10.1111/jcmm.13268. [Google Scholar] [PubMed] [CrossRef]
82. Kerkelä E, Saarialho-Kere U. Matrix metalloproteinases in tumor progression: focus on basal and squamous cell skin cancer. Exp Dermatol. 2003;12(2):109–25. doi:10.1034/j.1600-0625.2003.120201.x. [Google Scholar] [PubMed] [CrossRef]
83. Ruffini F, Graziani G, Levati L, Tentori L, D’Atri S, Lacal PM. Cilengitide downmodulates invasiveness and vasculogenic mimicry of neuropilin 1 expressing melanoma cells through the inhibition of αvβ5 integrin. Int J Cancer. 2015;136(6):E545–58. doi:10.1002/ijc.29252. [Google Scholar] [PubMed] [CrossRef]
84. Contois LW, Akalu A, Caron JM, Tweedie E, Cretu A, Henderson T, et al. Inhibition of tumor-associated αvβ3 integrin regulates the angiogenic switch by enhancing expression of IGFBP-4 leading to reduced melanoma growth and angiogenesis in vivo. Angiogenesis. 2015;18:31–46. doi:10.1007/s10456-014-9445-2. [Google Scholar] [PubMed] [CrossRef]
85. Brooks PC, Montgomery AMP, Rosenfeld M, Reisfeld RA, Hu TH, Klier G, et al. Integrin αvβ3 antagonists promote tumor-regression by inducing apoptosis of Angiogenic blood-vessels. Cell. 1994;79(7):1157–64. doi:10.1016/0092-8674(94)90007-8. [Google Scholar] [PubMed] [CrossRef]
86. Dome B, Raso E, Dobos J, Meszaros L, Varga N, Puskas LG, et al. Parallel expression of αIIbβ3 and αvβ3 integrins in human melanoma cells upregulates bFGF expression and promotes their angiogenic phenotype. Int J Cancer. 2005;116(1):27–35. doi:10.1002/ijc.20991. [Google Scholar] [PubMed] [CrossRef]
87. Cardones AR, Murakami T, Hwang ST. CXCR4 enhances adhesion of B16 tumor cells to endothelial cells in vitro and in vivo via β1 integrin. Cancer Res. 2003;63(20):6751–7. [Google Scholar] [PubMed]
88. Kato H, Liao ZJ, Mitsios JV, Wang HY, Deryugina EI, Varner JA, et al. The primacy of β1 integrin activation in the metastatic cascade. PLoS One. 2012;7(10):e46576. doi:10.1371/journal.pone.0046576. [Google Scholar] [PubMed] [CrossRef]
89. Liu YQ, Zou XM, Sun GR, Bao YH. Codonopsis lanceolata polysaccharide CLPS inhibits melanoma metastasis via regulating integrin signaling. Int J Biol Macromol. 2017;103:435–40. doi:10.1016/j.ijbiomac.2017.05.093. [Google Scholar] [PubMed] [CrossRef]
90. Cascone I, Napione L, Maniero F, Serini G, Bussolino F. Stable interaction between α5β1 integrin and Tie2 tyrosine kinase receptor regulates endothelial cell response to Ang-1. J Cell Biol. 2005;170(6):993–1004. doi:10.1083/jcb.200507082. [Google Scholar] [PubMed] [CrossRef]
91. Urabe F, Kosaka N, Ito K, Kimura T, Egawa S, Ochiya T. Extracellular vesicles as biomarkers and therapeutic targets for cancer. Am J Physiol Cell Physiol. 2020;318(1):C29–39. doi:10.1152/ajpcell.00280.2019. [Google Scholar] [PubMed] [CrossRef]
92. Möller A, Lobb RJ. The evolving translational potential of small extracellular vesicles in cancer. Nat Rev Cancer. 2020;20(12):697–709. doi:10.1038/s41568-020-00299-w. [Google Scholar] [PubMed] [CrossRef]
93. Hu T, Wolfram J, Srivastava S. Extracellular vesicles in cancer detection: hopes and hypes. Trends Cancer. 2021;7:122–33. doi:10.1016/j.trecan.2020.09.003. [Google Scholar] [PubMed] [CrossRef]
94. Welsh JA, Goberdhan DCI, O’Driscoll L, Buzas EI, Blenkiron C, Bussolati B, et al. Minimal information for studies of extracellular vesicles (MISEV2023from basic to advanced approaches. J Extracell Vesicles. 2024;13(2):e12404. doi:10.1002/jev2.12404. [Google Scholar] [PubMed] [CrossRef]
95. Wu WC, Song SJ, Zhang Y, Li X. Role of extracellular vesicles in autoimmune pathogenesis. Front Immunol. 2020;11:579043. doi:10.3389/fimmu.2020.579043. [Google Scholar] [PubMed] [CrossRef]
96. Sigdel S, Swenson S, Wang J. Extracellular vesicles in neurodegenerative diseases: an update. Int J Mol Sci. 2023;24(17):13161. doi:10.3390/ijms241713161. [Google Scholar] [PubMed] [CrossRef]
97. Gołębiewska JE, Wardowska A, Pietrowska M, Wojakowska A, Dębska-Ślizień A. Small extracellular vesicles in transplant rejection. Cells. 2021;10(11):2989. doi:10.3390/cells10112989. [Google Scholar] [PubMed] [CrossRef]
98. Zhang C, Qin C, Dewanjee S, Bhattacharya H, Chakraborty P, Jha NK, et al. Tumor-derived small extracellular vesicles in cancer invasion and metastasis: molecular mechanisms, and clinical significance. Mol Cancer. 2024;23(1):18. doi:10.1186/s12943-024-01932-0. [Google Scholar] [PubMed] [CrossRef]
99. Dixson AC, Dawson TR, Di Vizio D, Weaver AM. Context-specific regulation of extracellular vesicle biogenesis and cargo selection. Nat Rev Mol Cell Biol. 2023;24(7):454–76. doi:10.1038/s41580-023-00576-0. [Google Scholar] [PubMed] [CrossRef]
100. Gurung S, Perocheau D, Touramanidou L, Baruteau J. The exosome journey: from biogenesis to uptake and intracellular signalling. Cell Commun Signal. 2021;19(1):47. doi:10.1186/s12964-021-00730-1. [Google Scholar] [PubMed] [CrossRef]
101. Biagioni A, Laurenzana A, Menicacci B, Peppicelli S, Andreucci F, Bianchini S, et al. uPAR-expressing melanoma exosomes promote angiogenesis by VE-Cadherin, EGFR and uPAR overexpression and rise of ERK1,2 signaling in endothelial cells. Cell Mol Life Sci. 2021;78(6):3057–72. doi:10.1007/s00018-020-03707-4. [Google Scholar] [PubMed] [CrossRef]
102. Ekström EJ, Bergenfelz C, Von Bülow V, Serifler F, Carlemalm E, Jönsson G, et al. WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol Cancer. 2014;13:88. doi:10.1186/1476-4598-13-88. [Google Scholar] [PubMed] [CrossRef]
103. Hakulinen J, Sankkila L, Sugiyama N, Lehti K, Keski-Oja J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J Cell Biochem. 2008;105(5):1211–8. doi:10.1002/jcb.21923. [Google Scholar] [PubMed] [CrossRef]
104. Lima LG, Oliveira AS, Campos LC, Bonamino M, Chammas R, Werneck C, et al. Malignant transformation in melanocytes is associated with increased production of procoagulant microvesicles. Thromb Haemost. 2011;106(4):712–23. doi:10.1160/TH11-03-0143. [Google Scholar] [PubMed] [CrossRef]
105. Hood JL, Pan H, Lanza GM, Wickline SA. Consortium for Translational Research in Advanced Imaging and Nanomedicine (C-TRAIN). Paracrine induction of endothelium by tumor exosomes. Lab Invest. 2009;89(11):1317–28. doi:10.1038/labinvest.2009.94. [Google Scholar] [PubMed] [CrossRef]
106. Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–91. doi:10.1038/nm.2753. [Google Scholar] [PubMed] [CrossRef]
107. Kim J, Afshari A, Sengupta R, Sebastiano V, Gupta A, Kim YH. Reproducibility project: cancer biology. Replication study: melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. elife. 2018;7:e39944. doi:10.7554/eLife.39944. [Google Scholar] [PubMed] [CrossRef]
108. Hu T, Hu J. Melanoma-derived exosomes induce reprogramming fibroblasts into cancer-associated fibroblasts via Gm26809 delivery. Cell Cycle. 2019;18(22):3085–94. doi:10.1080/15384101.2019.1669380. [Google Scholar] [PubMed] [CrossRef]
109. Zhou X, Yan T, Huang C, Xu Z, Wang L, Jiang E, et al. Melanoma cell-secreted exosomal miR-155-5p induce proangiogenic switch of cancer-associated fibroblasts via SOCS1/JAK2/STAT3 signaling pathway. J Exp Clin Cancer Res. 2018;37(1):242. doi:10.1186/s13046-018-0911-3. [Google Scholar] [PubMed] [CrossRef]
110. Yeon JH, Jeong HE, Seo H, Cho S, Kim K, Na D, et al. Cancer-derived exosomes trigger endothelial to mesenchymal transition followed by the induction of cancer-associated fibroblasts. Acta Biomater. 2018;76:146–53. doi:10.1016/j.actbio.2018.07.001. [Google Scholar] [PubMed] [CrossRef]
111. Tang H, Zhou X, Zhao X, Luo X, Luo T, Chen Y, et al. HSP90/IKK-rich small extracellular vesicles activate pro-angiogenic melanoma-associated fibroblasts via the NF-kB/CXCL1 axis. Cancer Sci. 2022;113(4):1168–81. doi:10.1111/cas.15271. [Google Scholar] [PubMed] [CrossRef]
112. Zhou Q, Fang T, Wei S, Chai S, Yang H, Tao M, et al. Macrophages in melanoma: a double-edged sword and targeted therapy strategies (Review). Exp Ther Med. 2022;24(4):640. doi:10.3892/etm.2022.11577. [Google Scholar] [PubMed] [CrossRef]
113. Tian JW, Zhang HJ, Li SY, Guo YL, Chen G, Yu ZL. Tumor cell-derived extracellular vesicles in modulating phenotypes and immune functions of macrophages: mechanisms and therapeutic applications. J Cancer. 2023;14(8):1321–34. doi:10.7150/jca.84632. [Google Scholar] [PubMed] [CrossRef]
114. Hood JL. Melanoma exosomes enable tumor tolerance in lymph nodes. Med Hypotheses. 2016;90:11–3. doi:10.1016/j.mehy.2016.02.018. [Google Scholar] [PubMed] [CrossRef]
115. Jarosz-Biej M, Kamińska N, Matuszczak S, Cichoń T, Pamuła-Piłat J, Czapla J, et al. M1-like macrophages change tumor blood vessels and microenvironment in murine melanoma. PLoS One. 2018;13(1):e0191012. doi:10.1371/journal.pone.0191012. [Google Scholar] [PubMed] [CrossRef]
116. Parikh R, Parikh S, Berzin D, Vaknine H, Ovadia S, Likonen D, et al. Recycled melanoma-secreted melanosomes regulate tumor-associated macrophage diversification. EMBO J. 2024;43:3553–86. doi:10.1038/s44318-024-00103-7. [Google Scholar] [PubMed] [CrossRef]
117. García-Silva S, Benito-Martín A, Nogués L, Hernández-Barranco A, Mazariegos MS, Santos V, et al. Melanoma-derived small extracellular vesicles induce lymphangiogenesis and metastasis through an NGFR-dependent mechanism. Nat Cancer. 2021;2(12):1387–405. doi:10.1038/s43018-021-00272-y. [Google Scholar] [PubMed] [CrossRef]
118. Leary N, Walser S, He Y, Cousin N, Pereira P, Gallo A, et al. Melanoma-derived extracellular vesicles mediate lymphatic remodelling and impair tumour immunity in draining lymph nodes. J Extracell Vesicles. 2022;11(2):e12197. doi:10.1002/jev2.12197. [Google Scholar] [PubMed] [CrossRef]
119. Corrie PG, Marshall A, Nathan PD, Lorigan P, Gore M, Tahir S, et al. Adjuvant Bevacizumab for melanoma patients at high risk of recurrence: survival analysis of the AVAST-M trial. Ann Oncol. 2018;29:1843–52. doi:10.1093/annonc/mdy229. [Google Scholar] [PubMed] [CrossRef]
120. Yan X, Sheng X, Chi Z, Si L, Cui C, Kong Y, et al. Randomized phase II study of Bevacizumab in combination with Carboplatin plus Paclitaxel in patients with previously untreated advanced mucosal melanoma. J Clin Oncol. 2021;39(8):881–9. doi:10.1200/JCO.20.00902. [Google Scholar] [PubMed] [CrossRef]
121. Rahma OE, Tyan K, Giobbie-Hurder A, Brohl AS, Bedard PL, Renouf DJ, et al. Phase IB study of zivaflibercept plus pembrolizumab in patients with advanced solid tumors. J Immunother Cancer. 2022;10(3):e003569. doi:10.1136/jitc-2021-003569. [Google Scholar] [PubMed] [CrossRef]
122. Baginska J, Nau A, Gomez Diaz I, Giobbie-Hurder A, Weirather J, Vergara J, et al. Zivaflibercept plus pembrolizumab in patients with advanced melanoma resistant to anti-PD-1 treatment. Cancer Immunol Immunother. 2024;18,73(1):17. doi:10.1007/s00262-023-03593-2. [Google Scholar] [PubMed] [CrossRef]
123. Tarhini AA, Frankel P, Ruel C, Ernstoff MS, Kuzel TM, Logan TF, et al. NCI 8628: a randomized phase 2 study of ziv-aflibercept and high-dose interleukin 2 or high-dose interleukin 2 alone for inoperable stage III or IV melanoma. Cancer. 2018;124(22):4332–41. doi:10.1002/cncr.31734. [Google Scholar] [PubMed] [CrossRef]
124. Tarhini AA, Frankel P, Margolin KA, Christensen S, Ruel C, Shipe-Spotloe J, et al. Aflibercept (VEGF Trap) in inoperable stage III or stage IV melanoma of cutaneous or uveal origin. Clin Cancer Res. 2011;17(20):6574–81. doi:10.1158/1078-0432.CCR-11-1463. [Google Scholar] [PubMed] [CrossRef]
125. Hersey P, Sosman J, O’Day S, Richards J, Bedikian A, Gonzalez R, et al. A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin αvβ3, ± dacarbazine in patients with stage IV metastatic melanoma. Cancer. 2010;116(6):1526–34. doi:10.1002/cncr.24821. [Google Scholar] [PubMed] [CrossRef]
126. Moschos SJ, Sander CA, Wang W, Reppert SL, Drogowski LM, Jukic DM, et al. Pharmacodynamic (phase 0) study using etaracizumab in advanced melanoma. J Immunother. 2010;33(3):316–25. doi:10.1097/CJI.0b013e3181c1f216. [Google Scholar] [PubMed] [CrossRef]
127. Carvajal RD, Wong MK, Thompson JA, Gordon MS, Lewis KD, Pavlick AC, et al. A phase 2 randomised study of ramucirumab (IMC-1121B) with or without dacarbazine in patients with metastatic melanoma. Eur J Cancer. 2014;50(12):2099–107. doi:10.1016/j.ejca.2014.03.289. [Google Scholar] [PubMed] [CrossRef]
128. D’Angelo SP, Hamid OA, Tarhini A, Schadendorf D, Chmielowski B, Collichio FA, et al. A phase 2 study of Ontuxizumab, a monoclonal antibody targeting endosialin, in metastatic melanoma. Invest New Drugs. 2018;36:103–13. doi:10.1007/s10637-017-0530-4. [Google Scholar] [PubMed] [CrossRef]
129. Li S, Wu X, Yan X, Zhou L, Chi Z, Si L, et al. Toripalimab plus axitinib in patients with metastatic mucosal melanoma: 3-year survival update and biomarker analysis. J Immunother Cancer. 2022;10(2):e004036. doi:10.1136/jitc-2021-004036. [Google Scholar] [PubMed] [CrossRef]
130. Lian B, Li Z, Wu N, Li M, Chen X, Zheng H, et al. Phase II clinical trial of neoadjuvant anti-PD-1 (toripalimab) combined with axitinib in resectable mucosal melanoma. Ann Oncol. 2024;35(2):211–20. doi:10.1016/j.annonc.2023.10.793. [Google Scholar] [PubMed] [CrossRef]
131. Sheng X, Yan X, Chi Z, Si L, Cui C, Tang B, et al. Axitinib in combination with toripalimab, a humanized immunoglobulin G4 monoclonal antibody against programmed cell death-1, in patients with metastatic mucosal melanoma: an open-label phase IB trial. J Clin Oncol. 2019;37(32):2987–99. doi:10.1200/JCO.19.00210. [Google Scholar] [PubMed] [CrossRef]
132. Wei X, Mao L, Chi Z, Sheng X, Cui C, Kong Y, et al. Efficacy evaluation of Imatinib for the treatment of melanoma: evidence from a retrospective study. Oncol Res. 2019;27(4):495–501. doi:10.3727/096504018X15331163433914. [Google Scholar] [PubMed] [CrossRef]
133. Jung S, Armstrong E, Wei AZ, Ye F, Lee A, Carlino MS, et al. Clinical and genomic correlates of imatinib response in melanomas with KIT alterations. Br J Cancer. 2022;127(9):1726–32. doi:10.1038/s41416-022-01942-z. [Google Scholar] [PubMed] [CrossRef]
134. Stoff R, Asher N, Laks S, Steinberg Y, Schachter J, Shapira-Frommer R, et al. Real world evidence of Lenvatinib + anti PD-1 as an advanced line for metastatic melanoma. Front Oncol. 2023;13:1180988. doi:10.3389/fonc.2023.1180988. [Google Scholar] [PubMed] [CrossRef]
135. Ferrucci PF. Lenvatinib/Pembrolizumab as second line treatment for advanced melanoma patients refractory to programmed death 1 (PD-1)/programmed death ligand-1 (PD-L1) inhibitors. Ann Transl Med. 2023;11(8):296. doi:10.21037/atm-23-341. [Google Scholar] [PubMed] [CrossRef]
136. Arance A, de la Cruz-Merino L, Petrella TM, Jamal R, Ny L, Carneiro A, et al. Phase II LEAP-004 study of Lenvatinib plus Pembrolizumab for melanoma with confirmed progression on a programmed cell death protein-1 or programmed death ligand 1 inhibitor given as monotherapy or in combination. J Clin Oncol. 2023;1,41(1):75–85. doi:10.1200/JCO.22.00221. [Google Scholar] [PubMed] [CrossRef]
137. Dayer LE, Hutchins LF, Johnson JT. Treatment of metastatic melanoma with pazopanib: a report of five patient cases. J Oncol Pharm Pract. 2015;21(3):224–31. doi:10.1177/1078155214524084. [Google Scholar] [PubMed] [CrossRef]
138. Fruehauf JP, El-Masry M, Osann K, Parmakhtiar B, Yamamoto M, Jakowatz JG. Phase II study of pazopanib in combination with paclitaxel in patients with metastatic melanoma. Cancer Chemother Pharmacol. 2018;82(2):353–60. doi:10.1007/s00280-018-3624-6. [Google Scholar] [PubMed] [CrossRef]
139. Takeda T, Tsubaki M, Kato N, Genno S, Ichimura E, Enomoto A, et al. Sorafenib treatment of metastatic melanoma with c-Kit aberration reduces tumor growth and promotes survival. Oncol Lett. 2021;22(6):827. doi:10.3892/ol.2021.13089. [Google Scholar] [PubMed] [CrossRef]
140. Eisen T, Ahmad T, Flaherty KT, Gore M, Kaye S, Marais R, et al. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. Br J Cancer. 2006;95(5):581–6. doi:10.1038/sj.bjc.6603291. [Google Scholar] [PubMed] [CrossRef]
141. Tang F, Li S, Liu D, Chen J, Han C. Sorafenib sensitizes melanoma cells to vemurafenib through ferroptosis. Transl Cancer Res. 2020;9(3):1584–93. doi:10.21037/tcr.2020.01.62. [Google Scholar] [PubMed] [CrossRef]
142. Santini D, Vincenzi B, Venditti O, Dell’Aquila E, Frezza AM, Silletta M, et al. Sunitinib in malignant melanoma: a treatment option only for KIT-mutated patients? Future Oncol. 2013;9(12):1809–11. doi:10.2217/fon.13.170. [Google Scholar] [PubMed] [CrossRef]
143. Zeng F, Li Y, Meng Y, Sun H, He Y, Yin M, et al. BET inhibitors synergize with sunitinib in melanoma through GDF15 suppression. Exp Mol Med. 2023;55(2):364–76. doi:10.1038/s12276-023-00936-y. [Google Scholar] [PubMed] [CrossRef]
144. Cook N, Basu B, Biswas S, Kareclas P, Mann C, Palmer C, et al. A phase 2 study of vatalanib in metastatic melanoma patients. Eur J Cancer. 2010;46(15):2671–3. doi:10.1016/j.ejca.2010.07.014. [Google Scholar] [PubMed] [CrossRef]
145. Ko SY, Lee W, Kenny HA, Dang LH, Ellis LM, Jonasch E, et al. Cancer-derived small extracellular vesicles promote angiogenesis by heparin-bound, bevacizumab-insensitive VEGF, independent of vesicle uptake. Commun Biol. 2019;2:386. doi:10.1038/s42003-019-0609-x. [Google Scholar] [PubMed] [CrossRef]
146. Kim JH, Lee CH, Baek MC. Dissecting exosome inhibitors: therapeutic insights into small-molecule chemicals against cancer. Exp Mol Med. 2022;54(11):1833–43. doi:10.1038/s12276-022-00898-7. [Google Scholar] [PubMed] [CrossRef]
147. Tengler L, Schütz J, Tiedtke M, Jablonska J, Theodoraki MN, Nitschke K, et al. Plasma-derived small extracellular vesicles unleash the angiogenic potential in head and neck cancer patients. Mol Med. 2023;29(1):69. doi:10.1186/s10020-023-00659-w. [Google Scholar] [PubMed] [CrossRef]
148. Xia Y, Zhang J, Liu G, Wolfram J. Immunogenicity of extracellular vesicles. Adv Mater. 2024;36:e2403199. doi:10.1002/adma.202403199. [Google Scholar] [PubMed] [CrossRef]
149. Ghodasara A, Raza A, Wolfram J, Salomon C, Popat A. Clinical translation of extracellular vesicles. Adv Healthc Mater. 2023;12(28):e2301010. doi:10.1002/adhm.202301010. [Google Scholar] [PubMed] [CrossRef]
150. Sanz-Ros J, Mas-Bargues C, Romero-García N, Huete-Acevedo J, Dromant M, Borrás C. Extracellular vesicles as therapeutic resources in the clinical environment. Int J Mol Sci. 2023;24(3):2344. doi:10.3390/ijms24032344. [Google Scholar] [PubMed] [CrossRef]
151. Dai S, Wei D, Wu Z, Zhou X, Wei X, Huang H, et al. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol Ther J Am Soc Gene Ther. 2008;16:782–90. doi:10.1038/mt.2008.1. [Google Scholar] [PubMed] [CrossRef]
152. Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med. 2005;3:9. doi:10.1186/1479-5876-3-9. [Google Scholar] [PubMed] [CrossRef]
153. Escudier B, Dorval T, Chaput N, André F, Caby MP, Novault S, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of the first phase I clinical trial. J Transl Med. 2005;3:10. doi:10.1186/1479-5876-3-10. [Google Scholar] [PubMed] [CrossRef]
154. Pal A, Gori S, Yoo SW, Thomas AG, Wu Y, Friedman J, et al. Discovery of orally bioavailable and brain-penetrable prodrugs of the potent nSMase2 inhibitor DPTIP. J Med Chem. 2022;65(16):11111–25. doi:10.1021/acs.jmedchem.2c00562. [Google Scholar] [PubMed] [CrossRef]
155. Peng Y, Zhao M, Hu Y, Guo H, Zhang Y, Huang Y, et al. Blockade of exosome generation by GW4869 inhibits the education of M2 macrophages in prostate cancer. BMC Immunol. 2022;23(1):37. doi:10.1186/s12865-022-00514-3. [Google Scholar] [PubMed] [CrossRef]
156. Xiao W, Pahlavanneshan M, Eun CY, Zhang X, DeKalb C, Mahgoub B, et al. Matrix stiffness mediates pancreatic cancer chemoresistance through induction of exosome hypersecretion in a cancer associated fibroblasts-tumor organoid biomimetic model. Matrix Biol Plus. 2022;14:100111. doi:10.1016/j.mbplus.2022.100111. [Google Scholar] [PubMed] [CrossRef]
157. Auger C, Brunel A, Darbas T, Akil H, Perraud A, Bégaud G, et al. Extracellular vesicle measurements with nanoparticle tracking analysis: a different appreciation of up and down secretion. Int J Mol Sci. 2022;23(4):2310. doi:10.3390/ijms23042310. [Google Scholar] [PubMed] [CrossRef]
158. Chernova T, Grosso S, Sun XM, Tenor AR, Cabeza JZ, Craxton A, et al. Extracellular vesicles isolated from malignant mesothelioma cancer-associated fibroblasts induce pro-oncogenic changes in healthy mesothelial cells. Int J Mol Sci. 2022;23(20):12469. doi:10.3390/ijms232012469. [Google Scholar] [PubMed] [CrossRef]
159. Dautova Y, Kapustin AN, Pappert K, Epple M, Okkenhaug H, Cook SJ, et al. Calcium phosphate particles stimulate interleukin-1β release from human vascular smooth muscle cells: a role for spleen tyrosine kinase and exosome release. J Mol Cell Cardiol. 2018;115:82–93. doi:10.1016/j.yjmcc.2017.12.007. [Google Scholar] [PubMed] [CrossRef]
160. Ipinmoroti AO, Pandit R, Crenshaw BJ, Sims B, Matthews QL. Selective pharmacological inhibition alters human carcinoma lung cell-derived extracellular vesicle formation. Heliyon. 2023;9(6):e16655. doi:10.1016/j.heliyon.2023.e16655. [Google Scholar] [PubMed] [CrossRef]
161. Lee CH, Bae JH, Choe EJ, Park JM, Park SS, Cho HJ, et al. Macitentan improves antitumor immune responses by inhibiting the secretion of tumor-derived extracellular vesicle PD-L1. Theranostics. 2022;12(5):1971–87. doi:10.7150/thno.68864. [Google Scholar] [PubMed] [CrossRef]
162. Johansson L, Reyes JF, Ali T, Schätzl H, Gilch S, Hallbeck M. Lack of cellular prion protein causes Amyloid β accumulation, increased extracellular vesicle abundance, and changes to exosome biogenesis proteins. Mol Cell Biochem. 2024;136:41. doi:10.1007/s11010-024-05059-0. [Google Scholar] [PubMed] [CrossRef]
163. Shin JM, Lee CH, Son S, Kim CH, Lee JA, Ko H, et al. Sulfisoxazole elicits robust antitumour immune response along with immune checkpoint therapy by inhibiting exosomal PD-L1. Adv Sci. 2022;9(5):e2103245. doi:10.1002/advs.202103245. [Google Scholar] [PubMed] [CrossRef]
164. Mallaredy V, Roy R, Cheng Z, Thej C, Benedict C, Truongcao M, et al. Tipifarnib reduces extracellular vesicles and protects from heart failure. Circ Res. 2024;135(2):280–97. doi:10.1161/CIRCRESAHA.123.324110. [Google Scholar] [PubMed] [CrossRef]
165. Kumagai T, Shimogawara R, Ichimura K, Iwanaga S. Calpain inhibitor suppresses both extracellular vesicle-mediated secretion of miRNAs and egg production from paired adults of Schistosoma japonicum. Parasitol Int. 2022;87:102540. doi:10.1016/j.parint.2022.102540. [Google Scholar] [PubMed] [CrossRef]
166. Shimizu S, Tojima I, Nakamura K, Arai H, Kouzaki H, Shimizu T. Nasal polyp fibroblasts (NPFs)-derived exosomes are important for the release of vascular endothelial growth factor from cocultured eosinophils and NPFs. Auris Nasus Larynx. 2022;49(3):407–14. doi:10.1016/j.anl.2021.10.002. [Google Scholar] [PubMed] [CrossRef]
167. Ramírez-Ponce MP, Flores JA, Barrella L, Alés E. Ketotifen is a microglial stabilizer by inhibiting secretory vesicle acidification. Life Sci. 2023;319:121537. doi:10.1016/j.lfs.2023.121537. [Google Scholar] [PubMed] [CrossRef]
168. Federici C, Petrucci F, Caimi S, Cesolini A, Logozzi M, Borghi M, et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS One. 2014;9(2):e88193. doi:10.1371/journal.pone.0088193. [Google Scholar] [PubMed] [CrossRef]
169. Kosgodage US, Mould R, Henley AB, Nunn AV, Guy GW, Thomas EL, et al. Cannabidiol (CBD) is a novel inhibitor for exosome and microvesicle (EMV) release in cancer. Front Pharmacol. 2018;9:889. doi:10.3389/fphar.2018.00889. [Google Scholar] [PubMed] [CrossRef]
170. Kosgodage US, Trindade RP, Thompson PR, Inal JM, Lange S. Chloramidine/Bisindolylmaleimide-I-mediated inhibition of exosome and microvesicle release and enhanced efficacy of cancer chemotherapy. Int J Mol Sci. 2017;18(5):1007. doi:10.3390/ijms18051007. [Google Scholar] [PubMed] [CrossRef]
171. Bratengeier C, Johansson L, Liszka A, Bakker AD, Hallbeck M, Fahlgren A. Mechanical loading intensities affect the release of extracellular vesicles from mouse bone marrow-derived hematopoietic progenitor cells and change their osteoclast-modulating effect. FASEB J. 2024;38(1):e23323. doi:10.1096/fj.202301520R. [Google Scholar] [PubMed] [CrossRef]
172. Li M, Yu D, Williams KJ, Liu ML. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Arterioscler Thromb Vasc Biol. 2010;30(9):1818–24. doi:10.1161/ATVBAHA.110.209577. [Google Scholar] [PubMed] [CrossRef]
173. Li Y, Wang J, Chen S, Wu P, Xu S, Wang C, et al. miR-137 boosts the neuroprotective effect of endothelial progenitor cell-derived exosomes in oxyhemoglobin-treated SH-SY5Y cells partially via COX2/PGE2 pathway. Stem Cell Res Ther. 2020;11(1):330. doi:10.1186/s13287-020-01836-y. [Google Scholar] [PubMed] [CrossRef]
174. Gangadaran P, Ahn BC. Extracellular vesicle- and extracellular vesicle mimetics-based drug delivery systems: new perspectives, challenges, and clinical developments. Pharmaceutics. 2020;12(5):442. doi:10.3390/pharmaceutics12050442. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools