
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
GDF11 downregulates FOXP3 in T-cell acute lymphoblastic leukemia-derived cells and associates with restraining aggressiveness
1 Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, DCBS, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City, 09340, Mexico
2 Posgrado en Biología Experimental. DCBS, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City, 09340, Mexico
3 Departamento de Biología de la Reproducción, DCBS, Universidad Autónoma Metrolitana-Iztapalapa, Mexico City, 09340, Mexico
4 Laboratorio de Medicina Experimental, Unidad de Medicina Traslacional IIB/UNAM, Instituto Nacional de Cardiología Ignacio Chávez, Mexico City, 14080, Mexico
5 Laboratorio de Cardiología Traslacional, Unidad de Medicina Traslacional, IIB/UNAM, Instituto Nacional de Cardiología Ignacio Chávez, Mexico City, 14080, Mexico
6 Departamento de Enfermedades Infecciosas, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Mexico City, 14080, Mexico
* Corresponding Authors: ALEJANDRO ESCOBEDO-CALVARIO. Email: ; LUIS E. GOMEZ-QUIROZ. Email:
Oncology Research 2025, 33(8), 2075-2084. https://doi.org/10.32604/or.2025.064899
Received 26 February 2025; Accepted 22 May 2025; Issue published 18 July 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Growth differentiation factor 11 (GDF11), a transforming growth factor-beta superfamily member, is a crucial protein involved in many differentiation processes in embryogenesis and morphogenesis, and it has been extensively characterized due to its capacity to target poorly differentiated cells, including transformed or cancer cells. Aim: In the present work, we aimed to describe the effects on migration, proliferation, and metabolism in the T-cell acute lymphoblastic leukemia-derived cell line Jurkat. Methods: Based on previous evidence, we analyzed metabolic changes exerted by GDF11 and its relationship with the aggressive phenotype. Results: We found a profound impact on mitochondrial metabolism and reactive oxygen species content; these were related to a decrement in the expression of the transcription factor forkhead-box-protein P3 (FOXP3), which is highly involved in aggressiveness in leukemia cells; this was verified by a decrement in invasion capacity exhibited by the Jurkat cells under the GDF11 treatment. Conclusion: The results position the GDF11 response as a good alternative in the search for new therapeutic options for these diseases.Keywords
Abbreviations
| ALL | acute lymphoblastic leukemia |
| CD | cluster of differentiation |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DHE | dihydroethidium |
| FCCP | carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone |
| FBS | fetal bovine serum |
| FOXP3 | forkhead-box-protein P3 |
| GDF11 | growth differentiation factor 11 |
| OCR | oxygen consumption rate |
| OXPHOS | oxidative phosphorylation |
| PVDF | polyvinylidene difluoride membranes |
| ROS | reactive oxygen species |
| SEM | standard error of the mean |
| T-ALL | T-cell acute lymphoblastic leukemia |
| TGF-b | transforming growth factor-beta |
The growth differentiation factor 11 (GDF11) is a family member of the transforming growth factor-beta (TGF-b). It is reported to exert key activities in morphogenesis [1] but is also associated with muscle regeneration, targeting satellite cells [2], and protective properties in heat damage [3]. Although it has been involved in many controversies, we believe that those disagreements are, in part, due to the degree of differentiation of the target cell [4]. This is particularly relevant in cancer cells. Many reports have pointed out the effects of GDF11 on cancer [5]. Our group has characterized the antitumoral properties in liver cancer, decreasing proliferation, migration, clonogenicity capacity, spheroid formation [6], and a decrease in aberrant metabolism [7]; others have reported similar findings in other types of cancer, including breast [8], pancreas [9], and esophageal [10], among other solid tumors.
Considering that GDF11 impacts poorly differentiated cells, we decided to explore the effects of this growth factor on cells derived from human leukemia. There is poor evidence regarding the GDF11 in the biology of leukemia cells. Recently, Aslan and collaborators published that patients with acute myeloid leukemia presented increased serum levels of GDF11 [11], but this study was descriptive, providing only correlative evidence.
Hematological malignancy T-cell acute lymphoblastic leukemia (T-ALL) is a subtype of acute lymphoblastic leukemia (ALL) characterized by an overproduction of poorly differentiated T lymphocytes. It is the most common type of cancer in children [12]. T-ALL accounts for 10% to 15% of pediatric ALL cases and up to 25% of adults [13]. This leukemia is even more aggressive than B-cell ALL and is commonly associated with leukemic and lymphomatous manifestations [14]. There is much evidence for the biology studies of T-ALL and the use of the Jurkat cell line, mainly for apoptosis, T-cell activation, and signal transduction [15]; also, Jurkat cells express the transcription factor forkhead-box-protein P3 (FOXP3), which has been reported to control many pro-oncogenic properties conferring aggressiveness in cancer cells, and poor prognosis in patients [16]. It is implicated in T-cell regulation, activation, and differentiation and is considered a master regulator for developing and functioning CD4+/CD25+ regulatory T-cells [17].
In this research, we focused on characterizing the effects of GDF11 on the expression of FOXP3 and its implication in cancer cell aggressiveness.
Human recombinant Growth Differentiation Factor 11 (GDF11) was from Peprotech (Cat. 120-11, Rocky Hill, NJ, USA); as we previously reported, all treatments with the growth factor were at 50 ng/mL at different incubation times [6,7].
The Jurkat cell line was purchased from the American Type Culture Collection (ATCC, TIB-152, Manassas, VA, USA). Jurkat cell is an immortalized T lymphocyte cell line originally obtained from the peripheral blood of a patient with T-cell leukemia. Cells were cultured in RPMI 1640 medium (Gibco BRL, Cat. 31800022, New York, NY, USA). Culture used 10% fetal bovine serum (FBS, HyClone, Cat. SH3091003, Logan, UT, USA), 100 U/mL ampicillin, and 100 µg/mL streptomycin (Thermo Fisher Scientific, Cat. 15070063, Waltham, MA, USA). Cells were maintained at 37°C in a 5% CO2 and 90% humidity atmosphere. Cells were plated in plastic culture dishes (Corning, Cat. 353037, New York, NY, USA). The cell line was mycoplasma-free.
Protein isolation and quantification
Protein extraction was performed using M-PER (Thermo Fisher Scientific, Cat. 78505, Boston, MA, USA) lysis buffer containing protease inhibitors (Complete, Sigma-Aldrich, Cat. 11836170001) and phosphatase inhibitors (PhosSTOP, Sigma-Aldrich, Cat. 4906845001). Protein quantification was performed using the bicinchoninic acid (BCA) kit (Pierce, Thermo Fisher Scientific, BCA1-1KT, Waltham, MA, USA) according to the manufacturer’s instructions. We used 60 mg of protein in the Western blot assays.
Western blotting was performed following the protocol previously reported [6]. Briefly, proteins were loaded onto precast 4–20% gels (Invitrogen, Cat. WTG42012BOX, Waltham, MA, USA), transferred to polyvinylidene difluoride membranes (PVDF, Thermo Fisher Scientific, Cat. 88518), and probed with primary antibodies as listed in Table A1. The membranes were incubated with the corresponding horseradish peroxidase-conjugated secondary antibody. Immunoreactive bands were identified with ECL-Plus Western blotting substrate (Thermo Fisher Scientific Pierce, Cat. 32134). Images were analyzed using ImageJ v. 1.54p software (National Institutes of Health, Bethesda, MD, USA).
Cell viability was assayed by flow cytometry using the BD FACSCalibur flow cytometer (Becton Dickinson, Life Sciences, Franklin Lakes, NJ, USA); we used the LIVE/DEAD fixable green stain (Thermo Fisher Scientific, Cat. L23101) following the manufacturer’s instructions. Data were analyzed using FlowJoX (Version 10.X, Becton Dickinson Life Sciences).
Real-time cellular metabolism determination
The real-time oxygen consumption rate (OCR) was assayed with the Seahorse XFe24 Flux Analyzer (Agilent Technologies, Santa Clara, CA, USA), as we recently reported [18], and according to the manufacturer’s instructions. Briefly, 1 × 106 Jurkat cells were pre-treated using the recombinant human GDF11 every 24 h until 72 h in FBS media. After incubation, the cells were harvested immediately and replated at 175,000 cells/well in an XFe24-well plate format with a previous Cell-Tak (Corning) matrix treatment per well. We conducted the XF Cell Mito Stress Test (Agilent Technologies, Cat. 1030 15-100); cells were incubated with unbuffered assay media (XF Base RPMI Media with 10 mM glucose, 2 mM L-glutamine, and 1 mM pyruvate) followed by sequential injection of 0.5 μM oligomycin, 1 μM carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), and 0.5 μM antimycin A plus Rotenone. According to the previously referred protocol, OCR measurements were normalized with total cell protein.
Jurkat cells were centrifuged and incubated (37°C) in the dark with dihydroethidium (DHE, 5 μM, Invitrogen, D11347) using PBS for 15 min. Superoxide radicals were detected using a BD FACSCalibur (Becton Dickinson Life Science) flow cytometer. Excitation and emission wavelengths were 518 and 606 nm, respectively [19]. We acquired 10,000 events in duplicate. Data were analyzed using FlowJo™ Software (Becton Dickinson Life Sciences).
FOXP3 determination and nuclear colocalization
Cells were treated with GDF11 for 24, 48, and 72 h; after that, they were fixed using PFA (4%) and permeabilized using Cyto-Fast Fix/Perm buffer Set (BioLegend, Cat. 10482735, San Diego, CA, USA) on ice for 15 min. The anti-FOXP3 was applied according to the Table A1. The antibody was conjugated with phycoerythrin (PE). TGF-b (20 ng/mL for 24 h. Peprotech, Cat. 100-21,Cranbury, NJ, USA) was used as a positive control of FOXP3 expression. Flow cytometry analysis was performed using 10,000 events by duplicate in a BD FACSCalibur flow cytometer (Becton Dickinson Life Science), and data obtained were analyzed using FlowJoX (Becton Dickinson Life Science).
For confocal colocalization of FOXP3 in the nucleus, cells were treated at different times with GDF11; after that, cells were incubated with the mAb anti-FOXP3 (Thermo Fisher Scientific, Cat. 236A/E7) overnight, next day, cells were concentrated by centrifugation and incubated 5 min with 1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, Cat. D9542). Images and colocalization were determined using a multi-photon confocal microscope (Carl Zeiss LSM-780 NLO, Oberkochen, Germany) and the Zen Analyzer software (Carl Zeiss V3.11).
Cell proliferation was addressed by the colorimetric cell proliferation and cytotoxicity assay, Cell Counting Kit-8 (CCK-8; Sigma-Aldrich, Cat. 96992), following the manufacturer’s instructions. Treatments with 20 mM cisplatin, for 24 and 48 h, were used as a positive control for cell damage.
Jurkat cell invasion was assessed using Boyden’s chamber (Thermo Fisher Scientific, pore diameter 8 µm, Cat. 140629). We added 1 × 106 cells, treated or not with GDF11, in 600 µL of RPMI medium previously covered with 200 µL of Matrigel Matrix (Corning Matrigel, hESC-Qualified Matrix, Cat. CLS354277). The lower chamber had the same medium supplemented with 20% FBS. Chambers were incubated overnight, and we counted cells in the lower chamber. We used colchicine (Col, 10 mM, Sigma-Aldrich, Cat. C9754) as a negative control, and a chamber was Matrigel-untreated to observe the migratory process through the filter as a technical control (C).
Data are presented as mean ± SEM for at least three independent experiments; each experiment was conducted in triplicate. An analysis of variance (ANOVA) was used to compare the means of different groups, followed by multiple comparisons by the Tukey test. Graph Pad Prism software (Dotmatics, CA, USA) version 8 for macOS was used. Differences were considered statistically significant when p < 0.05.
GDF11 activates the canonical signaling pathway in Jurkat cells
First, to corroborate the specific response of Jurkat cells to GDF11, we treated cells with the growth factor at early times (5–60 min). We evaluated the activation of Smad proteins, the canonical signaling pathway in the TGF-β family. Phosphorylation of Smad2 and Smad3 was assessed by Western blot (Fig. 1A). The results show that both Smad3 (Fig. 1B) and Smad2 (Fig. 1C) were activated by GDF11, peaking at 5 min and progressively decreasing over time. We could not detect any effect on cell viability, as judged by the flow cytometry assay, at late incubation times (Fig. 1D,E).
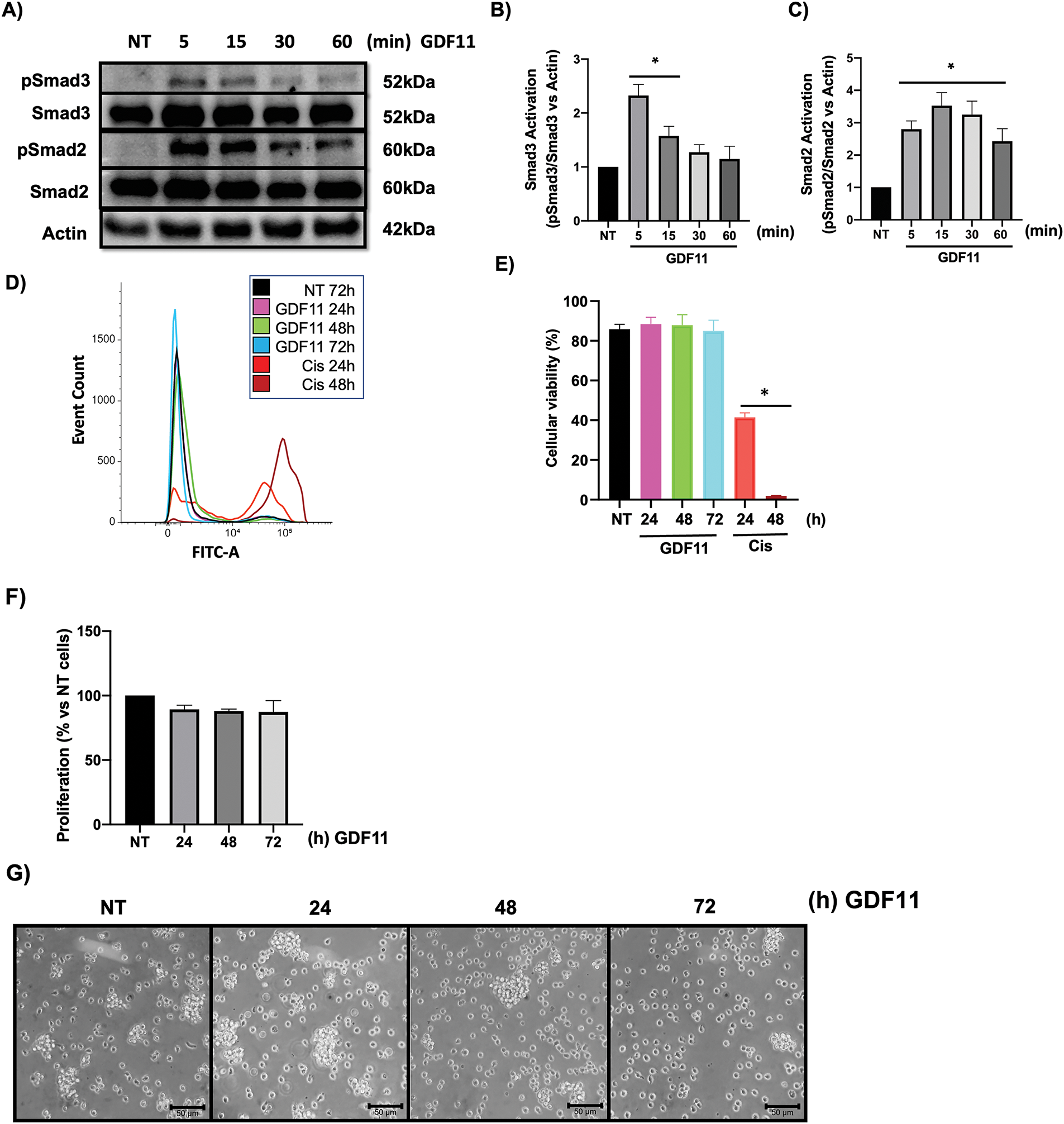
Figure 1: Jurkat cells respond to GDF11 treatment, activating Smad2 and Smad3 without affecting cell viability. Cells were treated with GDF11 (50 ng/mL) at various time points. (A) Western blot analysis of the Smad2 and Smad3 phosphorylation. Actin was used as a loading control. Densitometric analysis of (B) Smad3 and (C) Smad2. Each column represents the mean ± SEM of at least three independent experiments in triplicate. Cell viability was assessed by flow cytometry according to the protocol outlined in Materials and Methods. (D) Overlay histogram of the stained population; (E) Percentage of viable cells. Cisplatin (20 mM) at 24 and 48 h was used as a positive control; (F) Cell proliferation determined by CCK-8; Each column represents the mean ± SEM of at least three independent experiments in triplicate; (G) Cell morphology observed by light microscopy. Representative images of at least three independent experiments. *p < 0.05 vs. NT.
As previously reported, GDF11 has biological effects on cancer cells during prolonged incubation, particularly at 72 h; we treated cells for 24, 48, and 72 h. We did not observe changes in cell proliferation or morphology (Fig. 1F,G), which partially agrees with our previous results [6].
Metabolic effects induced by GDF11
Considering our previous findings in hepatocellular carcinoma cells, where we found changes in metabolic parameters, we performed a real-time metabolism study using the Seahorse technology [7]. We focused on mitochondria metabolism, exploring the oxygen consumption rate (OCR, Fig. 2A); we observed a clear separation in the bioenergetic behavior of the cells during the time of the study; profound data analysis revealed that treatment with GDF11 leads to metabolic modulation in the mitochondria, making more efficient the metabolism of these organelles, as indicated by basal respiration after 48 h (Fig. 2B), and maximal respiration at 48 and 72 h (Fig. 2C), reflected in the production of ATP at 72 h (Fig. 2D); and linked to an increment in non-mitochondrial oxygen consumption (Fig. 2E). These data demonstrate that GDF11 is involved in bioenergetics changes more than glycolytic metabolism, where we could not find any difference in Seahorse analysis (Data not shown).
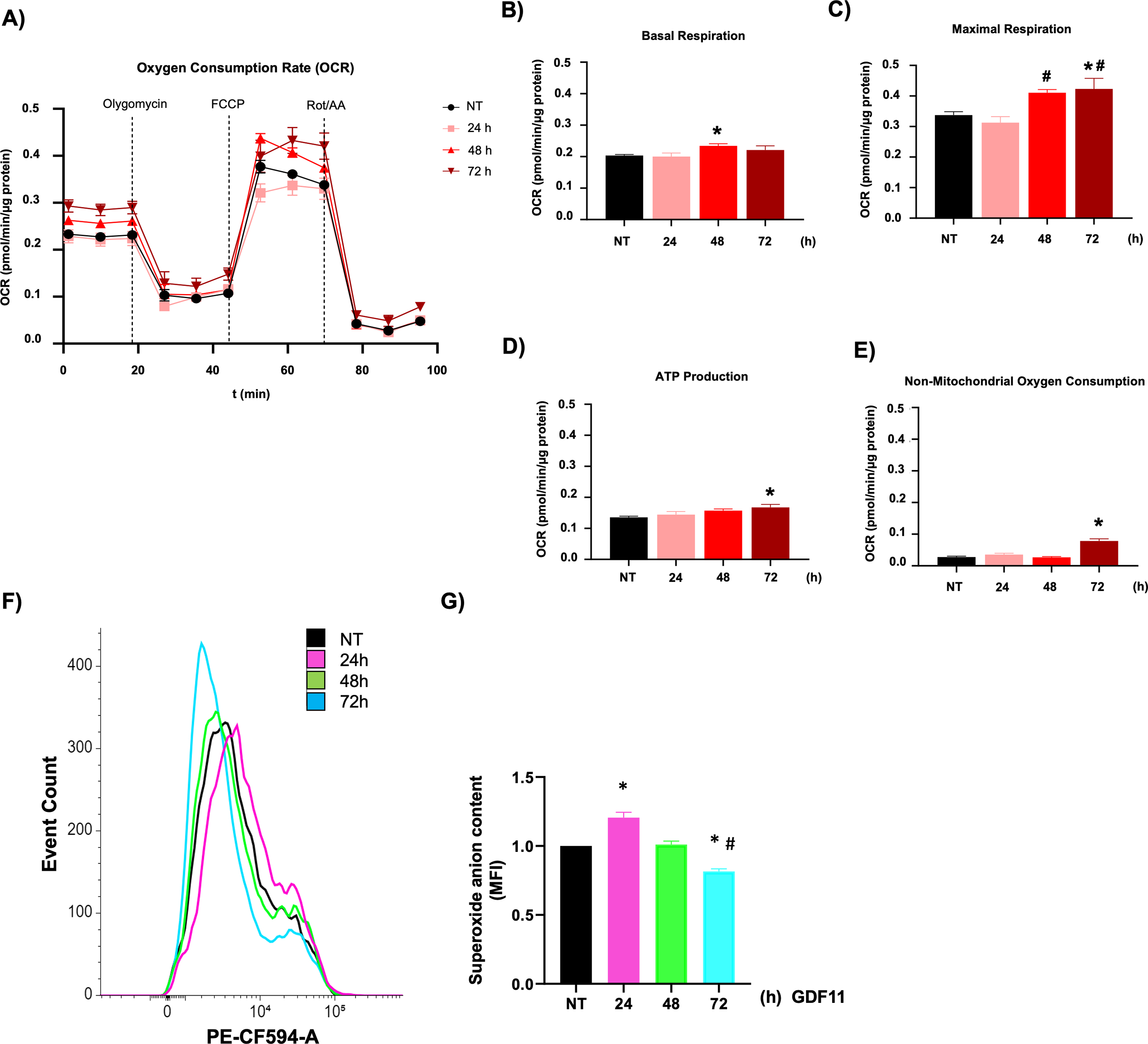
Figure 2: GDF11 promotes bioenergetic rewiring in Jurkat. (A) Mitochondrial metabolic activity was determined by oxygen consumption rate (OCR) using a Seahorse XF24e flux analyzer. The processed data were depicted as (B) basal respiration, (C) maximal respiration, (D) ATP production, and (E) non-mitochondrial oxygen consumption. (F) Overlay histogram of the DHE fluorescence; (G) The mean fluorescence intensity (MFI) of DHE. Each column represents the mean ± SEM of at least four independent experiments. *p < 0.05 vs. NT; #p < 0.05 vs. 24 h.
GDF11 modulates reactive oxygen species
The metabolism analysis suggests changes in the cell’s redox state. We analyzed the ROS content, observing a biphasic response of superoxide anion formation, increasing at 24 h of treatment and decreasing at 72 h, regarding untreated cells (Figs. 2F,G). The data indicate a possible signal modulation for different pathways in T cells, including mitochondria, led by ROS.
GDF11 decreases FOXP3 expression and reduces nuclear localization
It has been reported that ROS exerts a regulatory response in key transcription factors related to transformed T lymphocytes, such as FOXP3 [20,21]; the human protein atlas (proteinatlas.org) shows the high relevance of FOXP3 expression in different cancer cells, including leukemia (arrow, Fig. 3A), and Jurkat cells positions as a cell line with high expression of this transcription factor (arrow, Fig. 3B) [22].
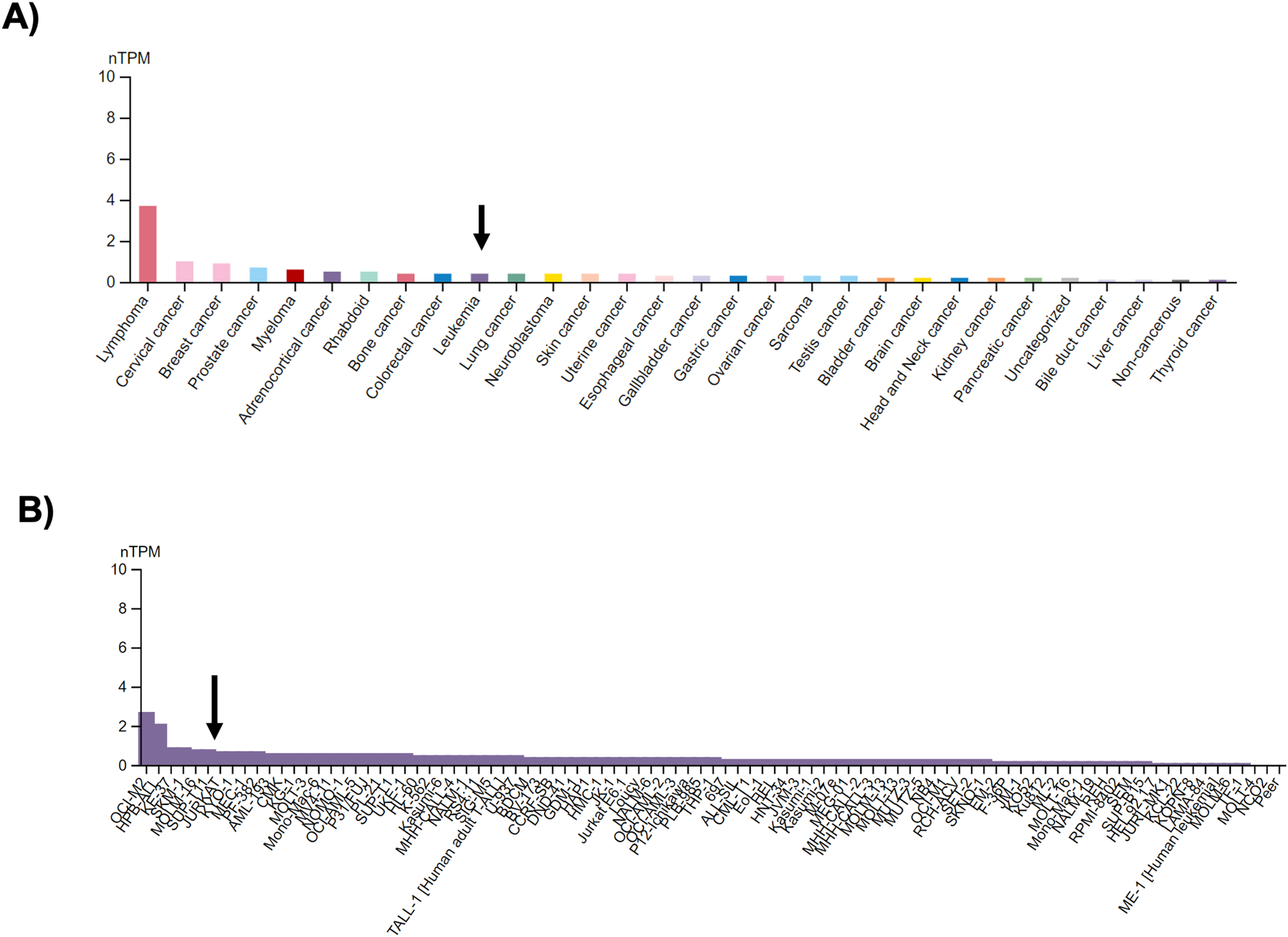
Figure 3: FOXP3 expression in cancer and human cancer-derived cell lines. (A) FOXP3 expression by cell category, according to the human protein atlas (proteinatlas.org). The arrow shows the expression of FOXP3 in Leukemia; and (B) FOXP3 expression in 93 leukemia cells, the arrow shows the Jurkat cell line. nTPM, normalized transcript per million.
Based on this evidence, we decided to explore the expression of this relevant protein in cells under GDF11 treatment. Fig. 4A,B shows that the FOXP3 content, determined by flow cytometry, follows a biphasic response, increasing at 24 h and decreasing to control values at 72 h; the TGF-b (20 ng/mL for 24 h) was used as a positive control of FOXP3 expression. This data strongly suggests a ROS-dependent regulation. To gain more confidence, we performed a colocalization study of FOXP3 in the cellular nucleus; Fig. 4C shows the basal colocalization of the transcription factor FOXP3 (anti-FOXP3, red) with the DNA (DAPI, blue), the treatment for 72 h with the GDF11 induces a decrease of the presence of FOXP3 in the nucleus. These data suggest a decrement in the content and nuclear localization of FOXP3 induced by the GDF11. FOXP3 is a critical transcription factor for leukemia cells; this protein is reportedly closely associated with high aggressiveness [23].
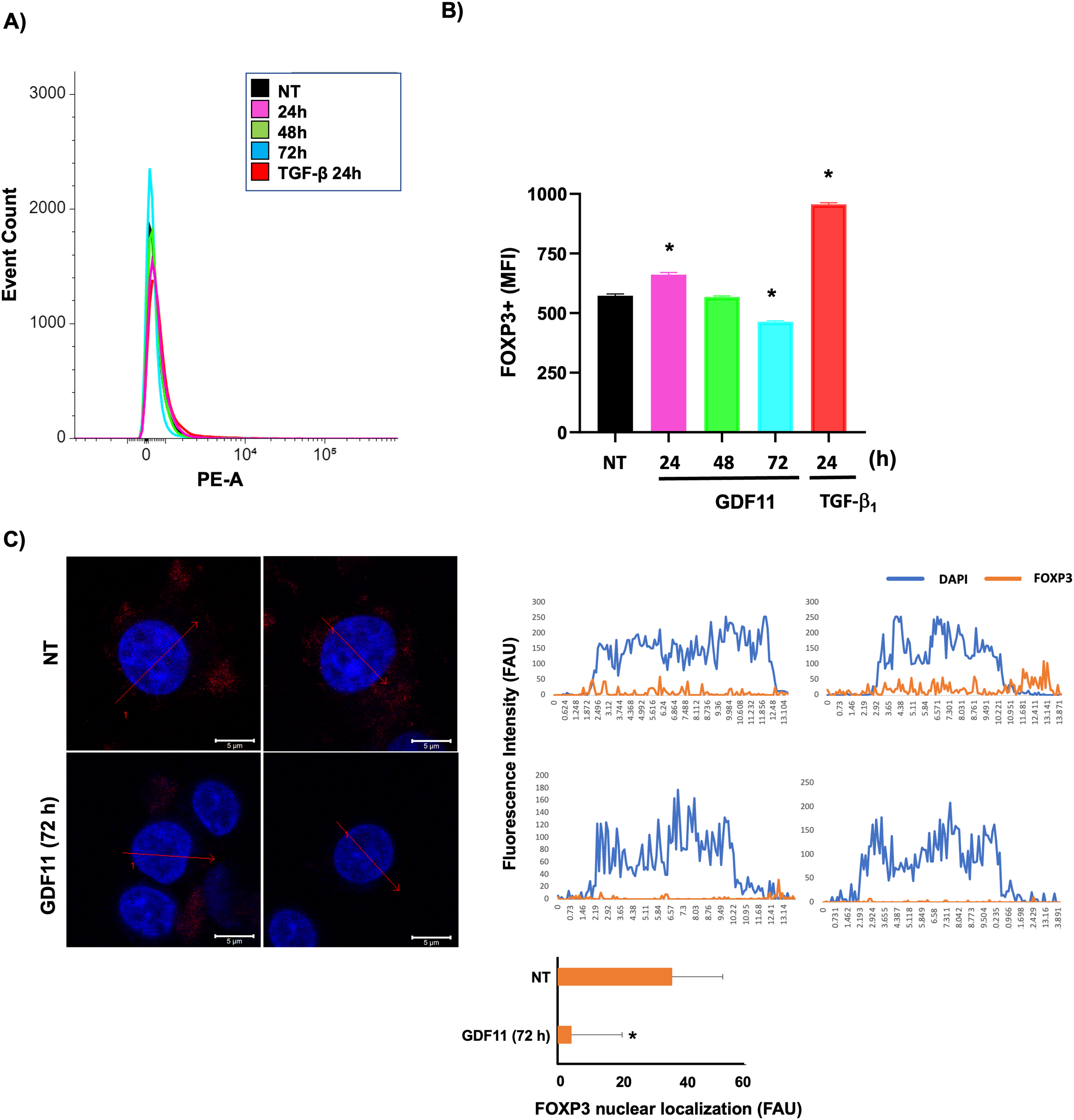
Figure 4: FOXP3 expression and nuclear localization in Jurkat cells. (A) Overlay histogram of the stained population; (B) The mean fluorescent intensity (MFI) of FOXP3-positive cells. (C) Nuclear co-localization of FOXP3 determined by confocal microscopy; intensity profiles of four different experiments of each cell group obtained with the Zen software, along a straight line (red arrow) crossing the nucleus of representative cells of FOXP3 (red) and DNA staining by DAPI (blue). The figure shows two representative images of each experiment. Histogram depicting the average quantification of fluorescence intensity plotted as fluorescence arbitrary units (FAU). *p < 0.05 vs. NT.
GDF11 limits migration capacity in Jurkat cells
To support the idea that GDF11 could induce a cellular response tending to decrease aggressivity in the leukemia cells, we assessed the migratory and invasive capacity of the cells under GDF11 treatment using Boyden’s chambers. Fig. 5A,B shows a time-dependent decrement in the migratory capacity, indicating a decrement in cell aggressiveness.
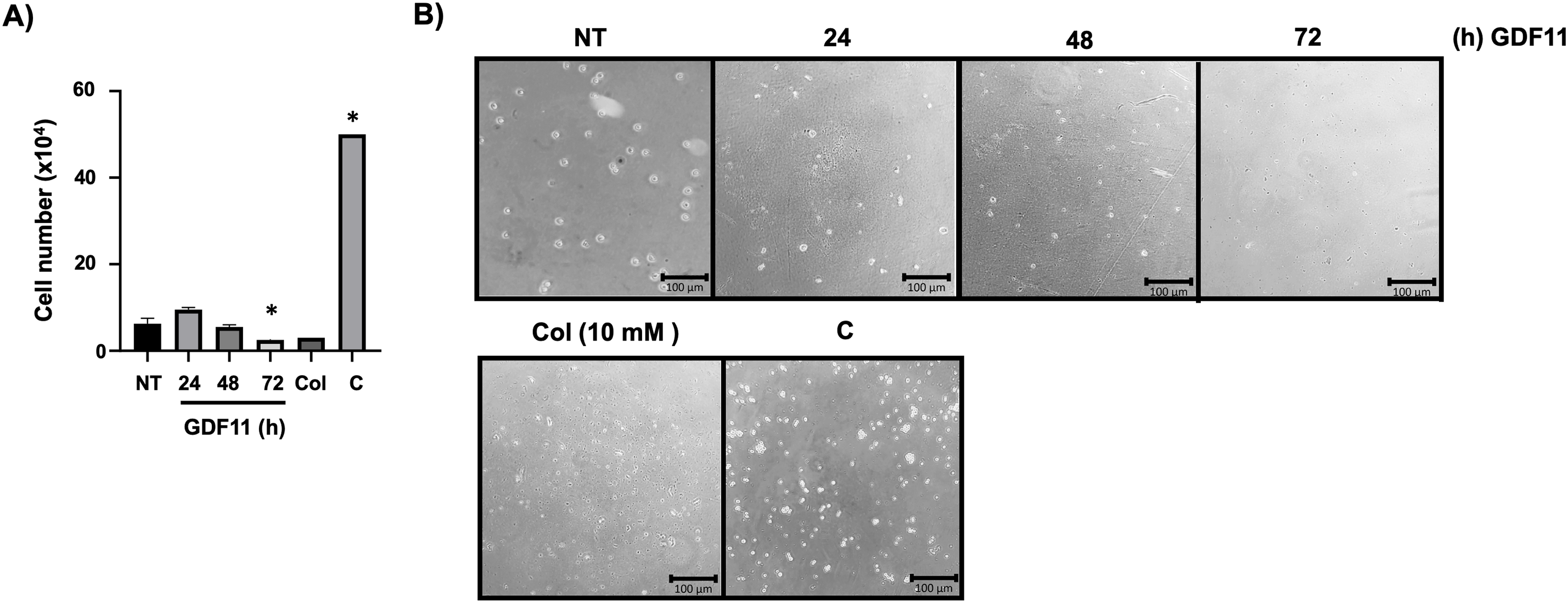
Figure 5: GDF11 limits migration and invasion capacity in Jurkat cells. (A) Migration and invasion assay determined by Boyden’s chambers. Cells were counted in the lower chamber at 24 h of the study. (B) Representative images of the cells in the lower chamber. Col (colchicine, 10 mM) as negative control, and (C) as technical control without Matrigel. Each column represents the mean ± SEM of at least three independent experiments in triplicate. *p < 0.05 vs. NT.
GDF11 has been implicated in many controversies, in part because foundational investigation in the biology of this growth factor was based on non-optimized research tools, for example, antibodies that could detect the GDF8 or myostatin, a close family member of the GDF11, and also because the degree of differentiation or stemness could be determinant in the biological response of the growth factor [4]. Considering these issues, exploring the effects of GDF11 in cancer biology could yield more insights.
It has been proposed that GDF11 can exert antitumoral effects in many cancer cell lines, including those from the liver [6,7], the breast [8], the esophagus [10], and the pancreas [9], among others; in fact, there is a clear association in survival in patients with high expression of GDF11 compared to those with low expression [5].
Previous works show that GDF11 exerts antitumoral effects in cancer cells, mainly targeting the aberrant metabolism of lipids and bioenergetics [7,24]. This was associated with the decrement of critical markers of aggressivity, such as spheroid formation, proliferation, clonogenicity capacity, migration, and mesenchymal to epithelial transition [6].
In the present study, we aimed to determine the relevance of GDF11 in T-cell acute lymphoblastic leukemia-derived cells using Jurkat cells. Due to the extensive use of this cell line, Jurkat cells serve as an effective model for exploring biological relevance, particularly regarding the significance of a critical disease like leukemia.
Our results demonstrated that Jurkat cells responded by activating the canonical signal transducers of Smad proteins, which agrees with many studies indicating the activation of Smad3 and Smad2 [6,24,25]. Proliferation and survival were not affected, but we found a profound change in mitochondrial metabolism, consistent with an anti-lipogenic response and a decrement in aggressiveness in cancer cells. It is reported that cancer cells can sequester mitochondria by overloading the organelle with cholesterol; this confers apoptosis resistance and aggressiveness but is detrimental to mitochondrial ATP production [26,27], to a more glycolytic metabolism.
Our data agree with these observations; we report an improvement in mitochondrial function judged by the Seahorse analysis of the OCR. Cruz and collaborators [28] show that oxidative phosphorylation (OXPHOS) is elevated in T-ALL cell lines, such as CCRF-CEM and Jurkat cells, compared to normal B and T cells. The basal OCR and ATP linked to respiration indicate mitochondrial dependence in T-ALL cells. Higher OCR from mitochondria represents more aggressiveness in leukemia cells due to a specific mechanism. The mitochondria constitutively transfer calcium from the endoplasmic reticulum through the inositol 1,4,5-triphosphate receptor (InsP3R), making all T-ALL bioenergetic cells susceptible to its inhibition. Cruz’s group used Xestospongin B (XeB), the InsP3R inhibitor; this reduced the bioenergetic parameters on T-ALL cells, inducing apoptosis, even though they are more effective on cell death when used in combination with dexamethasone [28]. We hypothesize that Jurkat cells increased OCR as a protective mechanism against cell death. However, this mechanism could indicate a double-edged sword; it may be more susceptible to combination therapies. Other reports using K562 and KCL-22, cells derived from chronic myelogenous and chronic myeloid leukemias, respectively, reported similar findings according to an increment in bioenergetics parameters such as OCR when using Imatinib, a BCR-ABL inhibitor. They reported reduced cell viability and apoptosis due to decreased caspase 3 activity; an increase in bioenergetic metabolism could have advantages and disadvantages [29].
Mitochondrial fitness could activate intrinsic apoptosis by releasing cytochrome c. Our results may suggest that GDF11 could have positive effects when combined with other therapies. On the other hand, mitochondria modulation could drive possible immunomodulatory or immunometabolic responses. Activation led by glycolysis could lead to Th1 (intracellular bacteria), Th2 (parasites), or Th17 (extracellular bacteria) activation in normal lymphocytes, contrary to OXPHOS, which carries out T regulatory (Treg) or memory cell phenotypes [30]. We can not specify a phenotype in the present study because Jurkat cells are not differentiated T cells. Still, we can hypothesize that they are associated with one specific secretome and specific markers such as FOXP3.
Interestingly, we observed a biphasic regulation of ROS content that correlated with the expression of FOXP3, a transcription factor influenced by ROS [21] and related to more aggressiveness [23]. Interestingly, according to The Cancer Protein Atlas, leukemia cells prefer an increment in FOXP3 expression compared to other types of cancer. The Jurkat cells represent one of the most aggressive ALL cell lines. There is evidence that the reduction of FOXP3 increases the survival rate in leukemia patients, and targeting this transcription factor could be a promising therapeutic strategy, even in solid tumors [17]. Our results show that GDF11 successfully decreased the expression of FOXP3, which was associated with a minor presence of the transcription factor in the nucleus. FOXP3 represents a malignant marker in non-cancerous and differentiated lymphocytes, such as Treg cells, that control and induce immunosuppressive properties in the tumor microenvironment [17,31].
A functional study of invasion addressed the impact of the aggressive phenotype; we demonstrated that GDF11-treated Jurkat cells significantly decreased the migratory and invasion capacity. It has been reported that Jurkat cells promote MMP-2 and MMP-9 secretion under pro-inflammatory stimuli [32,33].
Although this study provides evidence that GDF11 decreases some markers of aggressiveness in human-derived leukemia cells, it is important to note that the main limitation is the cellular model. This work presents an alternative approach to implementing pre-clinical studies that advance and consider the lessons learned from GDF11 as a negative regulator of FOXP3 in this type of cancer.
The present study provides evidence that GDF11 induces a decrease in the aggressiveness of Jurkat cells through a mechanism dependent on mitochondrial metabolic restoration and redox state modulation. This mechanism affects the expression and nuclear localization of the FOXP3 transcription factor. This research suggests that GDF11 could be an exciting target for further exploration in leukemia therapeutic approaches.
Acknowledgement: We thank the confocal microscopy core unit at Universidad Autónoma Metropolitana Iztapalapa.
Funding Statement: This work was partially funded by a grant from the Consejo Nacional de humanidades Ciencia y Tecnología (Conahcyt). Fronteras de la Ciencia 1320, Infra-2017 280788, and Universidad Autónoma Metropolitana-Iztapalapa.
Author Contributions: Melissa Sánchez-Rodríguez: Investigation, Methodology, Writing—original draft. Roberto Lazzarini-Lechuga: Investigation, Methodology. Verónica Souza-Arroyo: Resources, Visualization. Leticia Bucio-Ortiz: Supervision, Methodology. Roxana U. Miranda-Labra: Methodology. Monserrat Gerardo-Ramírez: Methodology, Visualization. Araceli Páez-Arenas: Sotfware. Moises Vergara-Mendoza: Methodology, Supervision. María Concepción Gutiérrez-Ruiz: Writing—original draft. Alejandro Escobedo-Calvario: Methodology, Writing—original draft. Luis E. Gomez-Quiroz: Funding acquisition, Resources, Writing—original draft. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The data supporting this study’s findings are available from the corresponding authors upon reasonable request by any qualified researcher.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
References
1. McPherron AC, Lawler AM, Lee SJ. Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat Genet. 1999;22(3):260–4. doi:10.1038/10320. [Google Scholar] [CrossRef]
2. Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014;344(6184):649–52. doi:10.1126/science.1251152. [Google Scholar] [CrossRef]
3. Chen L, Luo G, Liu Y, Lin H, Zheng C, Xie D, et al. Growth differentiation factor 11 attenuates cardiac ischemia reperfusion injury via enhancing mitochondrial biogenesis and telomerase activity. Cell Death Dis. 2021;12(7):665. doi:10.1038/s41419-021-03954-8. [Google Scholar] [CrossRef]
4. Gerardo-Ramirez M, German-Ramirez N, Escobedo-Calvario A, Chavez-Rodriguez L, Bucio-Ortiz L, Souza-Arroyo V, et al. The hepatic effects of GDF11 on health and disease. Biochimie. 2023;208:129–40. doi:10.1016/j.biochi.2022.12.017. [Google Scholar] [CrossRef]
5. Simoni-Nieves A, Gerardo-Ramirez M, Pedraza-Vazquez G, Chavez-Rodriguez L, Bucio L, Souza V, et al. GDF11 implications in cancer biology and metabolism. Facts and controversies. Front Oncol. 2019;9:1039. doi:10.3389/fonc.2019.01039. [Google Scholar] [CrossRef]
6. Gerardo-Ramirez M, Lazzarini-Lechuga R, Hernandez-Rizo S, Jimenez-Salazar JE, Simoni-Nieves A, Garcia-Ruiz C, et al. GDF11 exhibits tumor suppressive properties in hepatocellular carcinoma cells by restricting clonal expansion and invasion. Biochim Biophys Acta Mol Basis Dis. 2019;1865(6):1540–54. doi:10.1016/j.bbadis.2019.03.003. [Google Scholar] [CrossRef]
7. Hernandez S, Simoni-Nieves A, Gerardo-Ramirez M, Torres S, Fucho R, Gonzalez J, et al. GDF11 restricts aberrant lipogenesis and changes in mitochondrial structure and function in human hepatocellular carcinoma cells. J Cell Physiol. 2021;236(5):4076–90. doi:10.1002/jcp.30151. [Google Scholar] [CrossRef]
8. Bajikar SS, Wang CC, Borten MA, Pereira EJ, Atkins KA, Janes KA. Tumor-suppressor inactivation of GDF11 occurs by precursor sequestration in triple-negative breast cancer. Dev Cell. 2017;43(4):418–435 e13. doi:10.1016/j.devcel.2017.10.027. [Google Scholar] [CrossRef]
9. Liu Y, Shao L, Chen K, Wang Z, Wang J, Jing W, et al. GDF11 restrains tumor growth by promoting apoptosis in pancreatic cancer. Onco Targets Ther. 2018;11:8371–9. doi:10.2147/ott.s181792. [Google Scholar] [CrossRef]
10. Gu B, Wang Q, Dai W, Ma G, Song Y, Wu Q. Effect of GDF11 on proliferation and apoptosis of esophageal cancer cells. Cell Mol Biol. 2018;64(11):80–4. doi:10.14715/cmb/2018.64.11.14. [Google Scholar] [CrossRef]
11. Aslan NA, Avci E, Senol H, Guler N. EXPRESS: gDF11 level and its effect on prognosis in patients with acute myeloid leukemia. J Investig Med. 2024;72(4):341–8. doi:10.1177/10815589241238218. [Google Scholar] [CrossRef]
12. Girardi T, Vicente C, Cools J, De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood. 2017;129(9):1113–23. doi:10.1182/blood-2016-10-706465. [Google Scholar] [CrossRef]
13. Patel AA, Thomas J, Rojek AE, Stock W. Biology and treatment paradigms in T cell acute lymphoblastic leukemia in older adolescents and adults. Curr Treat Options Oncol. 2020;21(7):57. doi:10.1007/s11864-020-00757-5. [Google Scholar] [CrossRef]
14. Litzow MR, Ferrando AA. How I treat T-cell acute lymphoblastic leukemia in adults. Blood. 2015;126(7):833–41. doi:10.1182/blood-2014-10-551895. [Google Scholar] [CrossRef]
15. Gioia L, Siddique A, Head SR, Salomon DR, Su AI. A genome-wide survey of mutations in the Jurkat cell line. BMC Genomics. 2018;19(1):334. doi:10.1101/118117. [Google Scholar] [CrossRef]
16. Osborne DG, Domenico J, Fujita M. Expression of IL-37 induces a regulatory T-cell-like phenotype and function in Jurkat cells. Cells. 2022;11(16):2565. doi:10.3390/cells11162565. [Google Scholar] [CrossRef]
17. Roncador G, Garcia JF, Garcia JF, Maestre L, Lucas E, Menarguez J, et al. FOXP3, a selective marker for a subset of adult T-cell leukaemia/lymphoma. Leukemia. 2005;19(12):2247–53. doi:10.1038/sj.leu.2403965. [Google Scholar] [CrossRef]
18. Diaz-Basilio F, Vergara-Mendoza M, Romero-Rodriguez J, Hernandez-Rizo S, Escobedo-Calvario A, Fuentes-Romero LL, et al. The ecto-enzyme CD38 modulates CD4 T cell immunometabolic responses and participates in HIV pathogenesis. J Leukoc Biol. 2024;116(3):440–55. doi:10.1093/jleuko/qiae060. [Google Scholar] [CrossRef]
19. Enriquez-Cortina C, Almonte-Becerril M, Clavijo-Cornejo D, Palestino-Dominguez M, Bello-Monroy O, Nuno N, et al. Hepatocyte growth factor protects against isoniazid/rifampicin-induced oxidative liver damage. Toxicol Sci. 2013;135(1):26–36. doi:10.1093/toxsci/kft134. [Google Scholar] [CrossRef]
20. Shimojima Y, Kishida D, Ichikawa T, Takamatsu R, Nomura S, Sekijima Y. Oxidative stress promotes instability of regulatory T cells in antineutrophil cytoplasmic antibody-associated vasculitis. Front Immunol. 2021;12:789740. doi:10.3389/fimmu.2021.789740. [Google Scholar] [CrossRef]
21. Kim JE, Shin JS, Moon JH, Hong SW, Jung DJ, Kim JH, et al. Foxp3 is a key downstream regulator of p53-mediated cellular senescence. Oncogene. 2017;36(2):219–30. doi:10.1038/onc.2016.193. [Google Scholar] [CrossRef]
22. Uhlen M, Karlsson MJ, Hober A, Svensson AS, Scheffel J, Kotol D, et al. The human secretome. Sci Sig. 2019;12(609):1–8. doi:10.1126/scisignal.aaz0274. [Google Scholar] [CrossRef]
23. Karube K, Aoki R, Sugita Y, Yoshida S, Nomura Y, Shimizu K, et al. The relationship of FOXP3 expression and clinicopathological characteristics in adult T-cell leukemia/lymphoma. Mod Pathol. 2008;21(5):617–25. doi:10.1038/modpathol.2008.25. [Google Scholar] [CrossRef]
24. Luo H, Guo Y, Liu Y, Wang Y, Zheng R, Ban Y, et al. Growth differentiation factor 11 inhibits adipogenic differentiation by activating TGF-beta/Smad signalling pathway. Cell Prolif. 2019;52(4):e12631. doi:10.1111/cpr.12631. [Google Scholar] [CrossRef]
25. Honda M, Makino T, Zhao X, Matsuto M, Sakurai H, Takahashi Y, et al. Pathophysiological levels of GDF11 activate Smad2/Smad3 signaling and induce muscle atrophy in human iPSC-derived myocytes. Am J Physiol Cell Physiol. 2022;323(5):C1402–9. doi:10.1152/ajpcell.00341.2022. [Google Scholar] [CrossRef]
26. Zhang H, Xia N, Tang T, Nie S, Zha L, Zhang M, et al. Cholesterol suppresses human iTreg differentiation and nTreg function through mitochondria-related mechanisms. J Transl Med. 2023;21(1):224. doi:10.1186/s12967-023-03896-z. [Google Scholar] [CrossRef]
27. Garcia-Ruiz C, Conde de la Rosa L, Ribas V, Fernandez-Checa JC. Mitochondrial cholesterol and cancer. Semin Cancer Biol. 2021;73(1):76–85. doi:10.1016/j.semcancer.2020.07.014. [Google Scholar] [CrossRef]
28. Cruz P, Ahumada-Castro U, Bustos G, Molgo J, Sauma D, Lovy A, et al. Inhibition of InsP3R with Xestospongin B reduces mitochondrial respiration and induces selective cell death in T cell acute lymphoblastic leukemia cells. Int J Mol Sci. 2021;22(2):651. doi:10.3390/ijms22020651. [Google Scholar] [CrossRef]
29. De Rosa V, Monti M, Terlizzi C, Fonti R, Del Vecchio S, Iommelli F. Coordinate modulation of glycolytic enzymes and OXPHOS by imatinib in BCR-ABL driven chronic myelogenous leukemia cells. Int J Mol Sci. 2019;20(13):3134. doi:10.3390/ijms20133134. [Google Scholar] [CrossRef]
30. Angajala A, Lim S, Phillips JB, Kim JH, Yates C, You Z, et al. Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front Immunol. 2018;9:1605. doi:10.3389/fimmu.2018.01605. [Google Scholar] [CrossRef]
31. Mortezaee K. FOXP3 (in)stability and cancer immunotherapy. Cytokine. 2024;178(11):156589. doi:10.1016/j.cyto.2024.156589. [Google Scholar] [CrossRef]
32. Kossakowska AE, Edwards DR, Prusinkiewicz C, Zhang MC, Guo D, Urbanski SJ, et al. Interleukin-6 regulation of matrix metalloproteinase (MMP-2 and MMP-9) and tissue inhibitor of metalloproteinase (TIMP-1) expression in malignant non-Hodgkin’s lymphomas. Blood. 1999;94(6):2080–9. doi:10.1182/blood.v94.6.2080. [Google Scholar] [CrossRef]
33. Hajighasemi F. Profiles of MMP-2 expression in Jurkat, Molt-4 and U937 cells. Iran J Immunol. 2011;8(2):120–6. [Google Scholar]
Appendix A

Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools