
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Comparative Analysis of the Transcriptome and Metabolome in Leaves of Diploid and Tetraploid Fagopyrum tataricum
1 Key Laboratory of Coarse Cereal Processing, Ministry of Agriculture and Rural Affairs, School of Food and Biological Engineering, Chengdu University, Chengdu, 610106, China
2 Sichuan Provincial Academy of Natural Resource Sciences, Chengdu, 610015, China
3 School of Preclinical Medicine, Chengdu University, Chengdu, 610106, China
* Corresponding Authors: Xiaodong Shi. Email: ; Bei Niu. Email:
Phyton-International Journal of Experimental Botany 2023, 92(11), 3149-3162. https://doi.org/10.32604/phyton.2023.027324
Received 24 October 2022; Accepted 13 February 2023; Issue published 24 October 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Tartary buckwheat (Fagopyrum tataricum) is a dual-purpose medicinal and food crop grown for its high contents of functional compounds and abundant nutrients. Although studies have shown the differences of total flavonoid content in Tartary buckwheat at different ploidy levels, the composition of flavonoid and its regulatory mechanisms are largely unknown. In this study, the leaf metabolome and transcriptome of diploid and tetraploid accessions of Tartary buckwheat were analyzed to gain insight into the impact of polyploidization on comparative secondary metabolite composition and molecular regulatory mechanism. Based on a widely targeted metabolomics analysis, a total of 792 metabolites were identified, including 127 flavonoids. The accumulation of 127 metabolites and expression of 3871 genes differed significantly between diploid and tetraploid Tartary buckwheat. Integrated metabolomics and transcriptome analysis revealed that chromosome doubling up-regulated the expression of upstream genes in the flavonoid biosynthesis pathway to promote the accumulation of flavonoids. The present results contribute to elucidation of the molecular mechanism of phenotypic variation associated with polyploidy in Tartary buckwheat. The findings provide a reference for further studies on phenotypic traits in polyploid Tartary buckwheat, the cloning of crucial regulatory genes, and utilization of genetic engineering technologies in the breeding of Tartary buckwheat.Keywords
Tartary buckwheat (Fagopyrum tataricum Gaertn.), a member of the Polygonaceae family, is a popular food and medicinal crop with a bitter taste [1,2]. Tartary buckwheat is rich in starch, protein, dietary fiber, vitamins, minerals, and many other nutrients and metabolites [3,4]. In addition, Tartary buckwheat possesses multiple bioactive ingredients not shared by other seed and grain crops, such as flavonoids [5,6]. Tartary buckwheat is beneficial for prevention and treatment of high blood pressure, hyperglycemia, high blood lipid concentration, and cardiovascular disease, and for strengthening the immune system [7–9]. With the advantages of tolerance of drought and soil infertility, rapid growth, and strong adaptability, Tartary buckwheat is among the most important minor grain crops. It has become an important specialty crop of high nutritional and economic value, with broad prospective applications in health food development. At present, the seeds of buckwheat are the major edible site for human consumption, which has been widely used in a number of ways such as noodles, biscuits, liquor and tea [10]. Unfortunately, its hard shell and bitter taste inhibit its consumption [11]. Interestingly, the flavonoid content in flowers and leaves is higher than that in grains of buckwheat, which lay an excellent foundation for the development of whole plant [4,12]. For example, the material of whole plant Tartary buckwheat tea mainly includes buckwheat bran, flour, flower and stem. Besides Tartary buckwheat seeds, the whole plant will be valuable for its used in a wide range of applications in the food industries and pharmaceutical, especially the leaves [13]. In addition, leaf production is expected to become industrialized, as it is not limited by sites, climates and seasons than seed production. Therefore, buckwheat leaf and its production possess some unique properties, which are suitable for the development of new products.
In recent years, research on Tartary buckwheat has shown that polyploidy is a potent means of increasing crop yield and quality. After chromosome doubling, the size and thickness of leaf cells are increased [14]. However, the proportion of aborted pollen grains in tetraploid Tartary buckwheat is higher than that in diploids, indicating that the genetic stability of tetraploid Tartary buckwheat is lower than that of diploid accessions [15,16]. Lian et al. demonstrated that the average flavonoid and protein contents of tetraploid Tartary buckwheat were significantly higher than those of the diploid parents [17]. Chromosomal doubling in Tartary buckwheat influences certain quality traits. Previous studies have investigated autopolyploid induction and physiological differences in Tartary buckwheat [14–17]. Since polyploidy can increase the biomass of leaf, they are more suitable for large-scale production. However, comparison of the mechanism of gene regulation and phenotypic variation between tetraploid and diploid Tartary buckwheat has been rarely reported.
The development of high-throughput sequencing and bioinformatic technologies has enabled widespread application of transcriptome analysis to gain insights into the molecular mechanisms of plant metabolite biosynthesis [18,19]. In parallel, metabolomics (or metabonomics) is applied to understand the metabolic status and dynamic changes of a biological system, and has been increasingly applied to identify the crucial metabolites that are most directly associated with the phenotype of an organism [10–12]. The combination of multi-omic techniques can identify potential interactions between genes and metabolites, and explore molecular mechanisms and metabolic pathways [20]. For example, integrated metabolome and transcriptome tools have been applied to provide insights into the mechanisms of flavonoid accumulation in the exocarp of cucumber fruit [21]. Recently, multi-omic techniques have been extensively used in plant phenotyping analysis of many plant species, such as Camellia japonica, Manihot esculenta, and Carthamus tinctorius [22–24]. Consequently, the integration of transcriptome and metabolomic technologies provides a powerful tool for identification of genes and pathways associated with crucial biosynthetic mechanisms in plants. Furthermore, the method has been gradually recognized and widely used to investigate the biosynthesis of metabolites and the mechanisms of gene regulation in buckwheat [25,26]. In this study, a targeted ultra-high-performance liquid chromatography coupled with mass spectrometry (UHPLC-MS) metabolomics approach was applied to determine metabolic profiling and differential flavonoid metabolites among Tartary buckwheat accessions differing in ploidy. In addition, integration of metabolic and transcriptome analyses was conducted among the accessions with the aim of identifying the regulatory mechanisms at the metabolite and gene levels.
Tartary buckwheat seeds were obtained from the Key Laboratory of Coarse Cereal Processing, Chengdu University, China. Plants were grown in an experimental field and subjected to standard field management practices. Flow cytometry was used to analyze the DNA content in the leaf cells of diploid Tartary buckwheat and colchicine-induced materials to confirm that a stable tetraploid Tartary buckwheat material was obtained [27]. After about 20 days, the fifth leaf from the shoot apex of diploid and tetraploid Tartary buckwheat seedlings was sampled, flash frozen in liquid nitrogen, and stored at −80°C until use.
2.2 Sample Extraction and UHPLC-MS Analysis
The freeze-dried samples were crushed with a mixer mill for 30 s at 60 Hz. The ground samples were precisely weighed and transferred to an Eppendorf tube, after addition of 500 μL extract solution (methanol:water, 3:1 [v/v]). After vortexing, the samples were homogenized at 35 Hz for 4 min and sonicated for 5 min in an ice-water bath. After a series of treatments, the extract was stored at −80°C until the UHPLC-MS analysis. The UHPLC separation was conducted using an ExionLC System (SCIEX, Redwood City, CA, USA). The mobile phase consisted of solvent A (0.1% formic acid in water) and B (acetonitrile) by gradient elution. The separation was performed on a Waters Acquity UPLC HSS T3 (2.1 mm × 100 mm, 1.8 μm) with the column temperature was set at 40°C. Typical ion source parameters were as follows: Curtain Gas: 35 psi, Temperature: 400°C, Ion Source Gas 1:60 psi, Ion Source Gas 2: 60 psi, DP: ± 100 V.
2.3 Differential Metabolite Analysis
For metabolomics, SCIEX Analyst work station software (version 1.6.3) was employed for multiple reaction monitoring data acquisition and processing. After peak detection and annotation, the value of variable importance in the projection (VIP) of the first principal component in an orthogonal projections to latent structures discriminant analysis (OPLS-DA) was obtained. The VIP value summarizes the contribution of each variable to the model. The metabolites with VIP > 1 and p < 0.05 (Student’s t-test) were considered to be significantly changed metabolites. Principal component analysis (PCA) was performed using the Metware Cloud, a free online platform for data analysis (https://cloud.metware.cn). In addition, public databases, including Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/) and MetaboAnalyst (http://www.metaboanalyst.ca/), were accessed for pathway enrichment analysis. Metabolomic profiling was performed with the assistance of the Shanghai Biotree Biotech Co., Ltd. (Shanghai, China).
2.4 RNA Extraction and Sequencing
Total RNA was extracted using TRIzol reagent according to the manufacturer’s protocol. The RNA concentration and purity was measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). The RNA integrity was assessed using the RNA Nano 6000 Assay Kit and the Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). The PCR products were purified with the AMPure XP system and library quality was assessed with the Agilent Bioanalyzer 2100 system. The library preparations were sequenced on an Illuminaplatform and paired-end reads were generated. The sequencing data were uploaded to the NCBI database (BioProject ID: PRJNA833543). Gene expression levels were estimated as fragments per kilobase of transcript per million fragments mapped. Differential expression analysis of two groups was performed using DESeq2 software. The raw reads were processed with a bioinformatic pipeline tool on the BMKCloud online platform (http://www.biocloud.net).
3.1 Metabolic Profiling of Tartary Buckwheat Leaf
To obtain a comprehensive overview of the metabolite composition in the leaf and elucidate the metabolite differences between diploid and tetraploid Tartary buckwheat, a UHPLC-MS-based targeted metabolomic method was employed to screen the metabolic profile. The total ion current chromatograms of the different quality-control samples showed substantial overlap, which demonstrated the strong repeatability of the metabolite extraction and detection procedures (Supplementary Fig. S1). A total of 795 peaks were identified, and 792 metabolites were subjected to further analysis after recording of missing values and normalization. Lipids, nucleotides, amino acids, and derivatives comprised the major primary metabolites, followed by vitamins, carbohydrates, organic acids, and derivatives (Fig. 1a). The secondary metabolites were predominantly classified as flavonoids, alkaloids, terpenes, and phenylpropanoids. Flavonoids comprised the most abundant secondary metabolites. In total, 127 metabolites were identified as flavonoids, including six anthocyanins, seven chalcones, nine flavanols, eight flavanones, 43 flavones, 19 flavonols, and 16 isoflavones (Fig. 1b). The metabolite data were analyzed using PCA method to evaluate the overall variation among groups. The three biological replicates for each of the Diploid and tetraploid samples were clustered together, indicating that the results were highly reproducible among each group (Fig. 1c). The distance between the Diploid and tetraploid groups was large, reflecting metabolic differences with change in ploidy.
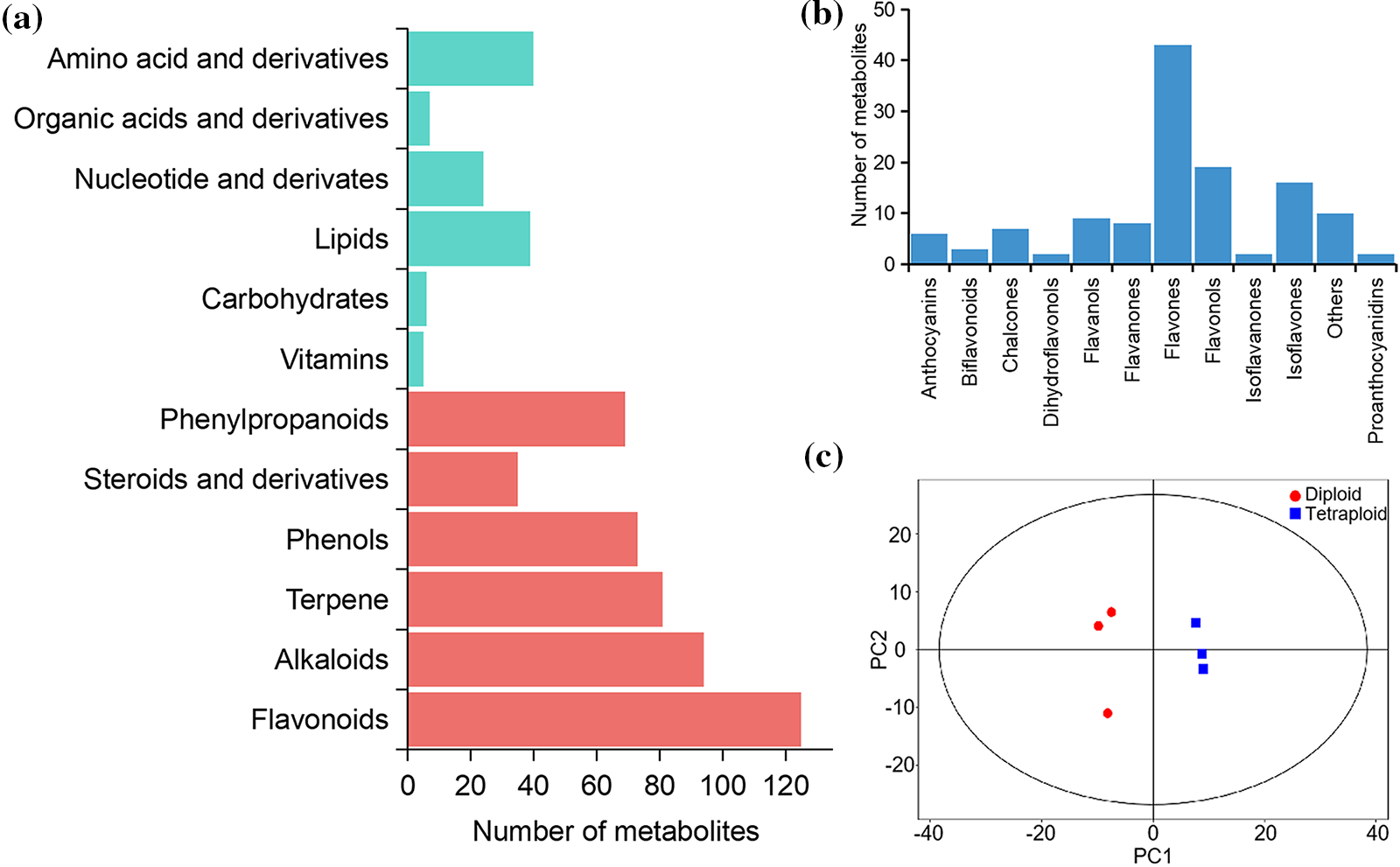
Figure 1: Classification of the annotated metabolites. (a) The amount of major metabolites detected. (b) Number of flavonoid metabolites identified. (c) PCA (Principal Component Analysis) score diagram of metabolic profiles in leaves
3.2 Identification of Differential Metabolites
To further identify differences in metabolites between the Diploid and tetraploid groups, supervised OPLS-DA models were generated. The Diploid and tetraploid groups formed distinct clusters in the OPLS-DA score plot. The model evaluation parameters R2X, R2Y, and Q2 were 0.479, 1.00, and 0.881, respectively. The parameter values indicated that the OPLS-DA model did not overfit the data, and showed strong discrimination and predictive power. Based on the OPLS-DA model, VIP < 1 and p < 0.05 (calculated with Student’s t-test) were applied as the criteria for screening differential metabolites (Fig. 2a). In total, 127 metabolites were differentially expressed between the Diploid and tetraploid groups, with 62 up-regulated and 65 down-regulated (Fig. 2b). Flavonoids were the most abundant differentially expressed metabolites, followed by alkaloids and then terpenes (Fig. 2c). The metabolic pathways of the differential metabolites were identified, based on a reference pathway library for the model plant Arabidopsis thaliana. The differential metabolites were mapped to 36 pathways, including metabolic pathways, biosynthesis of secondary metabolites, and ABC transporters. Based on annotations in the KEGG database, the pathways were involved in a large number of biosynthetic and metabolic processes, including 13 biosynthesis and 16 metabolism pathways. To determine whether the differentially expressed metabolites were significantly enriched with specific biological pathways, a pathway enrichment analysis was performed with MetaboAnalyst (Fig. 2d). The identified differential metabolites were predominantly enriched in pathways associated with amino acid metabolism and biosynthesis, such as alanine, aspartate, and glutamate metabolism, lysine biosynthesis, glycine, serine, and threonine metabolism, lysine degradation, arginine and proline metabolism, tyrosine metabolism, phenylalanine, tyrosine, and tryptophan biosynthesis, and tryptophan metabolism. In addition, phenylpropanoid biosynthesis, flavonoid biosynthesis, and flavone and flavonol biosynthesis were enriched among the differentially expressed flavonoids. These results indicated that metabolites accumulating in the leaf differed between diploid and tetraploid, and diverse primary and secondary metabolite pathways were significantly enriched among the differential metabolites.
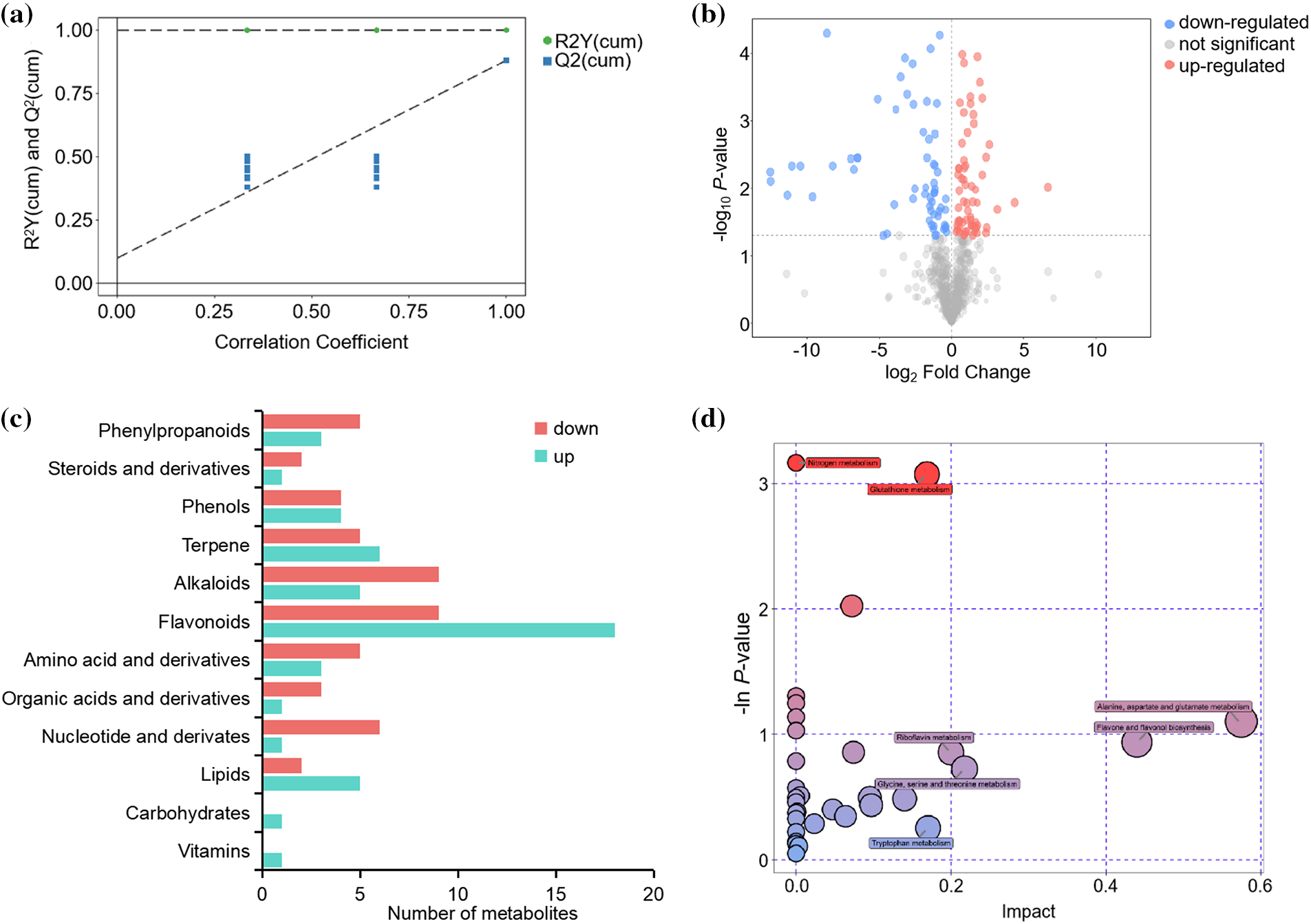
Figure 2: Analysis of differentially accumulated metabolites. (a) Differential metabolite analysis on the basis of OPLS-DA. (b) Volcano plot of metabolites. (c) The main categories of metabolites. (d) Differential metabolite KEGG enrichment. R2Y (cum) represents the interpretation rate of each model in Y axis direction in multivariate statistical analysis modeling, Q2 (cum) represents the prediction rate of each model
3.3 Analysis of Transcriptome Sequencing
To explore the molecular basis of the metabolic differences, the leaf transcriptome of diploid and tetraploid Tartary buckwheat was sequenced. Sequencing of six samples resulted in 36.69 Gb clean data after filtering. The Q30 base percentage was 90% or greater, indicating that the sequencing was reliable. Based on alignment with the Pinku1 reference genome, the alignment efficiency ranged from 86.75% to 96.70%. The correlation coefficients for the three biological replicates of each group were greater than 0.85, which satisfied the requirement for the following analysis. Overall, these results indicated that the transcriptome sequencing was of sufficient quality for further analysis. In total, 3871 differentially expressed genes (DEGs) were detected based on the screening criteria of fold change ≥ 2 and false discovery rate < 0.01. Among the DEGs, 1748 genes were up-regulated and 2123 genes were down-regulated. Of these genes, 1617 up-regulated and 1527 down-regulated genes were assigned functional annotations. Comparison of the Diploid and tetraploid groups revealed that up-regulated genes outnumbered down-regulated genes with chromosome doubling, and that a greater number of annotated genes were up-regulated than down-regulated.
A gene ontology (GO) functional analysis revealed that 2673 DEGs (1411 up-regulated and 1262 down-regulated) were classified to the three main GO categories (cellular component, molecular function, and biological process). In the molecular function category, DEGs were mainly associated with binding and catalytic activity. For the biological process and cellular component categories, major categories were metabolic process, cellular process, cell and membrane. Analysis of GO enrichment showed that the DEGs were significantly enriched in certain functional terms, of which the top 20 GO terms are presented in Fig. 3a. The GO enrichment analysis results for the cellular component category showed that more DEGs were enriched in GO terms associated with membranes, such as intrinsic component of membrane (GO:0031224), integral component of membrane (GO:0016021), membrane part (GO:0044425), and membrane (GO:0016020). With regard to the molecular function category, the DEGs were mostly enriched in terms associated with enzyme activity, such as oxidoreductase activity (GO:0016491), protein kinase activity (GO:0004672), kinase activity (GO:0016301), and transferase activity (GO:0016740). Interestingly, the significantly enriched terms in the biological process category were associated with cell wall formation, such as cell wall organization or biogenesis (GO:0071554), cellular polysaccharide metabolic process (GO:0044264), cellular polysaccharide biosynthetic process (GO:0033692), and cellular glucan metabolic process (GO:0006073).
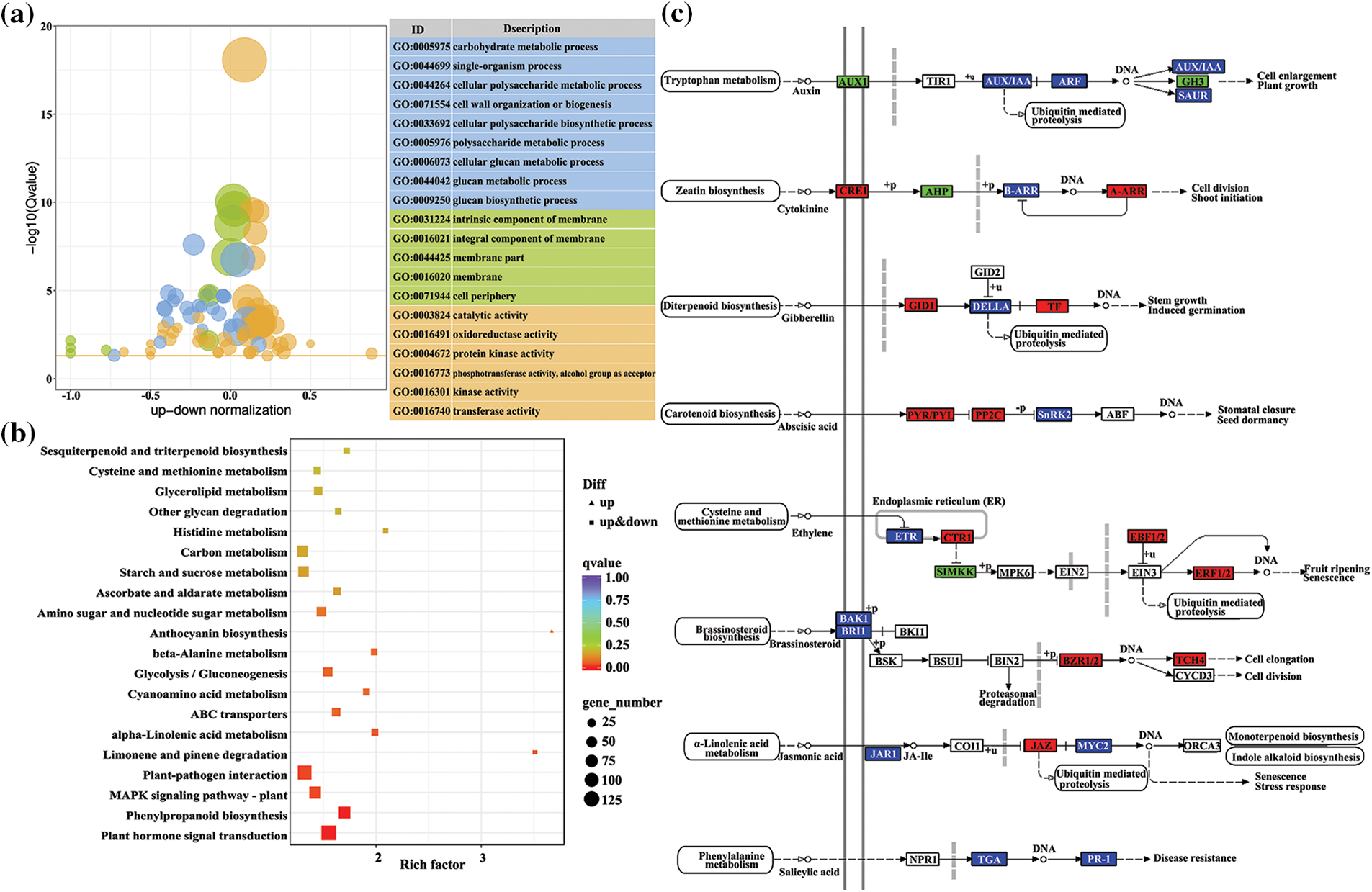
Figure 3: Top 20 enrichment GO term (a) and KEGG pathways (b) of DEGs. (c) DEGs on plant hormone signal transduction pathway
A KEGG pathway enrichment analysis revealed that most DEGs were mainly enriched in signaling pathways and in metabolic or biosynthetic pathways (Fig. 3b). The most significant enriched KEGG pathway was plant hormone signal transduction, followed by phenylpropanoid biosynthesis and MAPK signaling pathway. Therefore, plant hormone signal transduction and phenylpropanoid biosynthesis were selected for subsequent analysis. Phenylpropanoid biosynthesis was among the most important secondary metabolism pathways and is associated with the biosynthesis of flavonoids and lignin. A total of 132 DEGs were associated with plant hormone signal transduction, including auxin, cytokinin, gibberellin, abscisic acid, ethylene, brassinosteroids, jasmonic acid, and salicylic acid (Fig. 3c). Genes encoding for the cytokinin receptor CRE1, two-component response regulator ARR-A family, gibberellin receptor GID1, phytochrome interacting factor PIF3, abscisic acid receptor PYR/PYL family, protein phosphatase 2C, serine/threonine-protein kinase CTR1, ein3-binding F-box protein EBF1/2, ethylene-responsive transcription factor ERF1/2, brassinosteroid resistant BZR1/2, xyloglucosyl transferase TCH4, and jasmonate ZIM domain-containing protein JAZ were up-regulated, whereas auxin influx carrier AUX1, auxin responsive GH3, histidine-containing phosphotransfer protein AHP and mitogen-activated protein kinase kinase MKK were down-regulated. A signal network analysis indicated that genes in pathways upstream of plant hormone signaling were differentially expressed. These pathways included phenylalanine metabolism, brassinosteroid biosynthesis, cysteine and methionine metabolism, tryptophan metabolism, alpha-linolenic acid metabolism, carotenoid biosynthesis, diterpenoid biosynthesis, and zeatin biosynthesis. Genes involved in linolenic acid metabolism were up-regulated in the leaves of tetraploids, which in turn promoted the accumulation of jasmonic acid. In contrast, genes encoding UGT (UDP-glucosyltransferase) in the cytokinin signaling pathway were down-regulated in tetraploids. These results indicated that chromosome doubling in Tartary buckwheat may influence developmental processes and flavonoid biosynthesis by modulating the expression of genes involved in plant hormone signaling pathways and phenylpropanoid biosynthesis.
3.4 Integrated Analysis of the Transcriptome and Metabolome on the KEGG Pathway
Flavonoids are among the most important active metabolites in Tartary buckwheat. Based on the results of the widely targeted metabolomic analysis, the differential metabolites were mapped to flavonoid biosynthesis pathways, including flavonoid biosynthesis, flavone and flavonol biosynthesis, isoflavonoid biosynthesis, and anthocyanin biosynthesis (Fig. 4). In addition, the DEGs were indicated on the integrated metabolic map, thus the metabolites and genes were simultaneously mapped to pathways to clarify the relationships among flavonoid-related genes. Compared with the diploid group, the up-regulated genes associated with flavonoid formation were more numerous than the down-regulated genes in the tetraploid group. This result indicated that a greater number of flavonoid compounds were induced by these up-regulated genes and led to the significantly increased accumulation of the compounds. The integrated metabolic map suggested that chromosome doubling might up-regulate the expression of upstream genes in the flavonoid biosynthesis pathway (such as FtCHS and FtCHI) to promote the accumulation of flavonoids, such as cosmosiin, vitexin, and isovitexin. In contrast, the accumulation of several flavonoid compounds (e.g., nictoflorin and garbanzol) was decreased, which was suggestive of a reciprocal influence. Interestingly, in tetraploids, the quercetin content was significantly increased, whereas the quercitrin content decreased markedly, which may reflect the influence of other regulators between the two metabolites.
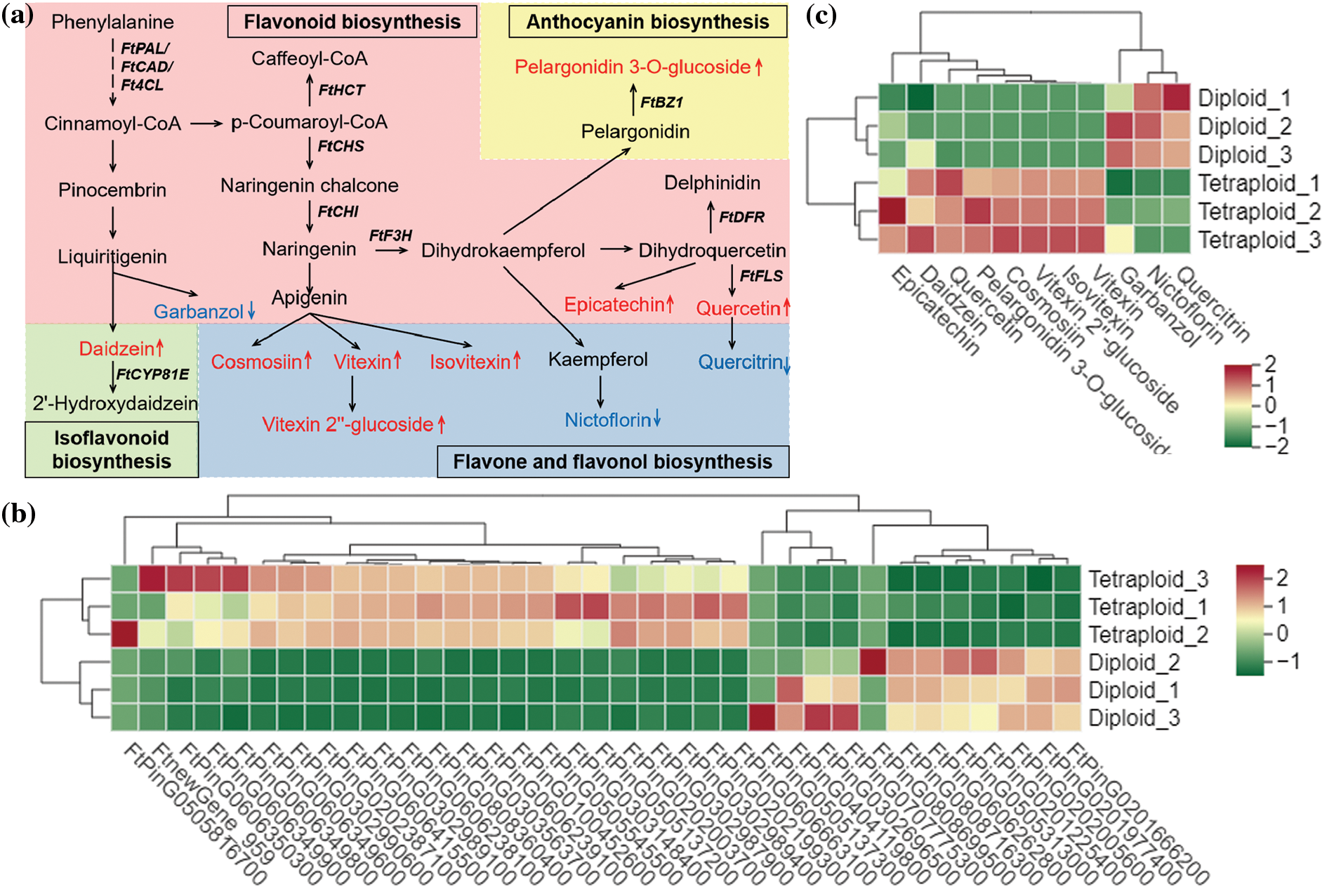
Figure 4: Simplified scheme (a) and heat map of the expression of genes (b) and metabolite (c) related to flavonoid biosynthesis. Red represented up-regulated metabolites and Blue represented down-regulated metabolites
Whole-genome duplication or polyploidy is considered to be an important driver of genomic evolution and may provide novel germplasm resources for plant improvement [28–30]. Although chromosome doubling does not directly introduce new genetic materials, the number of copies of an allele at a specific locus increases, ultimately leading to altered gene expression and phenotypic changes [31,32]. Relative to diploid plants, tetraploid plants show enhanced morphological variation. In polyploid plants, organ size is often increased and such plants generally have the advantage of enhanced stress tolerance. Production of polyploid forms has been widely applied in plant breeding [29,33,34]. However, the mechanisms for the phenotypic advantages of polyploid crops requires further investigation. In particular, transcriptome analysis can reveal the molecular mechanism of plant growth and development as well as various metabolic processes that will aid in understanding gene expression and the mining of crucial genes. In recent years, transcriptome and metabolome have played an important role in many areas of buckwheat active compositions, especially comparative metabolomics. Wang et al. successfully used GC-MS metabolomics for investigation of difference expressed metabolites in Tartary buckwheat cultivars M18 and JQ2, providing an effective method for the comprehensive assessment of cereal quality [11]. Yang et al. analyzed the relationship between secondary metabolites and their morphological variations using UPLC-MS-based metabolomics, which provided a reference for the precise breeding of new cultivars [25]. Based on the metabolomics at Tartary and common buckwheat flowers, a total of 188 flavonoid metabolites were detected [12]. High-throughput RNA-sequencing provides information on differences in gene expression patterns for searches of candidate genes or related pathways in polyploids [35–38]. In addition, metabolites are the basis of, and directly reflect, the plant phenotype, and allow an improved understanding of biological processes and mechanisms more intuitively and efficiently. Thus, integration of transcriptomic and metabolomic analyses was conducted to analyze the DEGs associated with metabolite accumulation and construct a core regulator network for different ploidies. Combination of metabolomic and transcriptomic data previously provided extensive characterization of the difference in invasive ability between two cytotypes of Solidago Canadensis [39]. A comparative transcriptomic and metabolomic study of Isatis indigotica on an autotetraploid and its progenitor was performed to construct a gene–metabolite correlation network [40]. Comparative metabolomics provide new information regarding the metabolites from various parts of the buckwheat plants, and offer important theoretical support for further comprehensive development and utilization of buckwheat. In this present study, we investigated the metabolic profiles in Tartary buckwheat leaves using the widely targeted metabolomics approach. A total of 127 metabolites were identified as flavonoids, laying the foundation for the development of buckwheat leaves. The present study used metabolomic and transcriptomic data to analyze differences in metabolite accumulation and gene expression in the leaves between diploid and autotetraploid Tartary buckwheat.
The phenotypic and physiological changes observed is considered to be predominantly the results of variation in gene expression. Thus, polyploids may significantly increase the potential variation in gene expression, which is reflected in phenotypic variation [41]. For example, stress-related genes are up-regulated after chromosome doubling for adaptation to environmental stress [42–44]. Many previous studies have indicated that phytohormone synthesis and signal transduction are an important cause of morphological differences between plants of different ploidies. In mulberry (Morus alba), the DEGs affect auxin and ethylene biosynthesis and signal transduction, and as a result tetraploids have superior phenotypic traits to diploids [45] Auxin-, cytokinin-, and brassinosteroid-related genes are up-regulated, causing significantly improved growth potential, in triploid poplar (Populus cathayana) [46]. In the present study, a large number of genes were enriched in plant signal transduction pathways, such as cytokinin receptor histidine kinases, gibberellin receptors, and ethylene response factors, which not only participate in plant hormone biosynthesis but also are involved in signal transduction. Many previous studies of plant hormone regulation of stress-tolerance genes have focused on analysis of plant adaptability to the external environment through polyploidization [44,47,48]. However, the stress tolerance of polyploids is not always consistent with a gene dosage effect, with more complex mechanisms underlying chromosome doubling [49]. Many studies have confirmed that genomic doubling is associated with variation in epigenetic modifications, such as DNA methylation and histone modifications [50–54]. Whether polyploidy and epigenetic modifications are correlated in Tartary buckwheat requires further research.
Polyploidization can affect metabolite biosynthesis with a profound impact on specific secondary metabolites [55,56]. Therefore, enhanced synthesis of phytochemical compounds through artificial chromosome doubling has been intensively investigated. Autotetraploid plants of Cichorium intybus accumulated higher contents of phenolic compounds than control plants [57]. In colchicine-induced polyploid Artemisia annua, tetraploid plants have increased artemisinin contents compared with that of diploid plants [58]. Although chromosome doubling-induced accumulation of active ingredients in medicinal plants is widely recognized, some species may present exceptions [56]. Tartary buckwheat is an important minor grain crop and is progressively becoming a model system to study the regulatory mechanism of flavonoid biosynthesis. Previous research has demonstrated that the expansion of key genes involved in secondary metabolite biosynthesis can be beneficial to the synthesis and accumulation of bioactive components [59]. A similar result was found in golden buckwheat (Fagopyrum dibotrys or Fagopyrum cymosum), in which the multiple duplicate genes were associated with flavonoid biosynthesis [26]. Some gene families essential for flavonoid biosynthesis, including FtCHS and FtCHI, were up-regulated in tetraploid Tartary buckwheat. And whole genome doubling has repercussions that reverberate throughout the transcriptome and downstream, ultimately generating altered the metabolite levels. The concentration of secondary metabolites in plants may be affected by phytohormone contents as well as endogenous physiological changes. Thus, the use of polyploid material in conjunction with exogenous hormone application may be an effective strategy for improving the accumulation of targeted metabolites. Future research will aim to develop an approach using a hormone-mediated system (e.g., hairy root or cell suspension) based on polyploid germplasm resources to produce valuable flavonoids, for example in Tartary buckwheat. This provides further evidence that the expansion of these structural and regulatory genes may be responsible for the increased flavonoid accumulation in tetraploid Tartary buckwheat.
In this study, transcriptome and metabolome analyses were conducted to investigate the metabolic differences between diploid and tetraploid Tartary buckwheat, with particular focus on the flavonoid biosynthetic pathway. Applying an UHPLC-MS procedure, 127 flavonoid metabolites were detected, of which 27 metabolites were significantly different. KEGG pathway enrichment analysis revealed that diverse primary and secondary metabolite pathways were significantly enriched. Chromosome doubling altered the expression of certain genes (such as genes involved in plant hormone signaling and the phenylpropanoid biosynthesis pathway) that ultimately led to changes in metabolite accumulation. Integrated metabolomics and transcriptome analysis showed that polyploidization up-regulated the expression of upstream genes in the flavonoid biosynthesis pathway, thus promoting flavone biosynthesis. However, the responses of other regulators and fine regulatory networks to chromosome doubling require further exploration.
Acknowledgement: There is no acknowledgement.
Funding Statement: This work was funded by National Key R&D Program of China (2019YFD1001300, 2019YFD1001303), the Earmarked Fund for China Agriculture Research System (CARS-08-02A) and the Opening Project of Key Laboratory of Coarse Cereal Processing of Ministry of Agriculture and Rural Affairs, Chengdu University (No. 2018CC5).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Xiaodong Shi, Bei Niu; data collection: Xiaodong Shi, Yue Qi; analysis and interpretation of results: Yue Qi, Jia Wang; draft manuscript preparation: Liangzhu Lin, Xiaobo Qin. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary Materials.
Ethics Approval: There is no applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Huang, J., Deng, J., Shi, T., Chen, Q., Liang, C. et al. (2017). Global transcriptome analysis and identification of genes involved in nutrients accumulation during seed development of rice tartary buckwheat (Fagopyrum Tararicum). Scientific Reports, 7(1), 11792. https://doi.org/10.1038/s41598-017-11929-z [Google Scholar] [PubMed] [CrossRef]
2. Zhang, K., He, M., Fan, Y., Zhao, H., Gao, B. et al. (2021). Resequencing of global Tartary buckwheat accessions reveals multiple domestication events and key loci associated with agronomic traits. Genome Biology, 22(1), 23. https://doi.org/10.1186/s13059-020-02217-7 [Google Scholar] [PubMed] [CrossRef]
3. Bonafaccia, G., Marocchini, M., Kreft, I. (2003). Composition and technological properties of the, flour and bran from common and tartary buckwheat. Food Chemistry, 80(1), 9–15. https://doi.org/10.1016/S0308-8146(02)00228-5 [Google Scholar] [CrossRef]
4. Zielinska, D., Turemko, M., Kwiatkowski, J., Zielinski, H. (2012). Evaluation of flavonoid contents and antioxidant capacity of the aerial parts of common and tartary buckwheat plants. Molecules, 17(8), 9668–9682. https://doi.org/10.3390/molecules17089668 [Google Scholar] [PubMed] [CrossRef]
5. Fabjan, N., Rode, J., Kosir, I. J., Wang, Z. H., Zhang, Z. et al. (2003). Tartary buckwheat (Fagopyrum tataricum Gaertn.) as a source of dietary rutin and quercitrin. Journal of Agricultural and Food Chemistry, 51(22), 6452–6455. https://doi.org/10.1021/jf034543e [Google Scholar] [PubMed] [CrossRef]
6. Joshi, D. C., Zhang, K., Wang, C., Chandora, R., Khurshid, M. et al. (2020). Strategic enhancement of genetic gain for nutraceutical development in buckwheat: A genomics-driven perspective. Biotechnology Advances, 39(6), 107479. https://doi.org/10.1016/j.biotechadv.2019.107479 [Google Scholar] [PubMed] [CrossRef]
7. Hu, Y., Hou, Z., Liu, D., Yang, X. (2015). Hypoglycemic and hepatoprotective effects of D-chiro-inositol-enriched tartary buckwheat extract in high fructose-fed mice. Food Function, 6(12), 3760–3769. https://doi.org/10.1039/C5FO00612K [Google Scholar] [PubMed] [CrossRef]
8. Hu, Y., Zhao, Y., Ren, D., Guo, J., Luo, Y. et al. (2016). Tartary buckwheat flavonoids protect hepatic cells against high glucose-induced oxidative stress and insulin resistance via MAPK signaling pathways. Food Function, 7(3), 1523–1536. https://doi.org/10.1039/C5FO01467K [Google Scholar] [PubMed] [CrossRef]
9. Lee, M. S., Shin, Y., Jung, S., Kim, S. Y., Jo, Y. H. et al. (2017). The inhibitory effect of tartary buckwheat extracts on adipogenesis and inflammatory response. Molecules, 22(7), 1160. https://doi.org/10.3390/molecules22071160 [Google Scholar] [PubMed] [CrossRef]
10. Li, L., Kong, Z. Y., Huang, X. J., Liu, Y. J., Liu, Y. J. et al. (2021). Transcriptomics integrated with widely targeted metabolomics reveals the mechanism underlying grain color formation in wheat at the grain-filling stage. Frontiers in Plant Science, 12, 757750. https://doi.org/10.3389/fpls.2021.757750 [Google Scholar] [PubMed] [CrossRef]
11. Wang, Y., Guan, Z. X., Liang, C. G., Liao, K., Xiang, D. B. et al. (2022). Agronomic and metabolomics analysis of rice-tartary buckwheat (Fagopyrum tataricum Gaertn) bred by hybridization. Scientific Reports, 12(1), 11986. https://doi.org/10.1038/s41598-022-16001-z [Google Scholar] [PubMed] [CrossRef]
12. Li, J., Hossain, M. S., Ma, H. C., Yang, Q. H., Gong, X. W. et al. (2020). Comparative metabolomics reveals differences in flavonoid metabolites among different coloured buckwheat flowers. Journal of Food Composition and Analysis, 85(10), 103335. https://doi.org/10.1016/j.jfca.2019.103335 [Google Scholar] [CrossRef]
13. Shi, X. D., Ru, H., Lin, L. Z., Wang, X. Y., Li, Y. J. et al. (2022). Comprehensive characterization in different types of tartary buckwheat tea based on intelligent sensory technology. Food Science and Technology, 42(4), e27222. https://doi.org/10.1590/fst.27222 [Google Scholar] [CrossRef]
14. Zhao, G., Tang, Y. (1994). Comparative study on main characters of a new strain of autotetraploid and its autodiploid parent stock in tartary buckwheat. Bulletin of Science and Technology, 10(5), 321–325. https://doi.org/10.13774/j.cnki.kjtb.1994.05.013 [Google Scholar] [CrossRef]
15. Wu, Y., Chen, Q. (2001). A cytological study on meiosis of PMCs of diploid and tetraploid tartary buckwheat. Guihaia, 21(4), 344–346. [Google Scholar]
16. Zhang, Y., He, P., Luo, Q., Wu, X., Kong, D. et al. (2020). Grain-filling of diploid and tetraploid tartary buckwheat. Fujian Journal of Agricultural Sciences, 35(10), 1043–1049. https://doi.org/10.19303/j.issn.1008-0384.2020.10.001 [Google Scholar] [CrossRef]
17. Lian, L., Chen, Q. (2013). A comparative study of seed protein content and seed flavonoid content between diploid and tetraploid tartary buckwheat. Seed, 32(2), 1–5. https://doi.org/10.16590/j.cnki.1001-4705.2013.02.051 [Google Scholar] [CrossRef]
18. Rai, A., Yamazaki, M., Takahashi, H., Nakamura, M., Kojoma, M. et al. (2016). RNA-seq transcriptome analysis of Panax japonicus, and its comparison with other panax species to identify potential genes involved in the saponins biosynthesis. Frontiers in Plant Science, 7, 481. https://doi.org/10.3389/fpls.2016.00481 [Google Scholar] [PubMed] [CrossRef]
19. Guo, X., Yu, C., Luo, L., Wan, H., Zhen, N. et al. (2017). Transcriptome of the floral transition in Rosa chinensis ‘Old Blush’. BMC Genomics, 18(1), 199. https://doi.org/10.1186/s12864-017-3584-y [Google Scholar] [PubMed] [CrossRef]
20. Bao, Y., Nie, T., Wang, D., Chen, Q. (2022). Anthocyanin regulatory networks in Solanum tuberosum L. leaves elucidated via integrated metabolomics, transcriptomics, and StAN1 overexpression. BMC Plant Biology, 22(1), 228. https://doi.org/10.1186/s12870-022-03557-1 [Google Scholar] [PubMed] [CrossRef]
21. Chen, C., Zhou, G., Chen, J., Liu, X., Lu, X. et al. (2021). Integrated metabolome and transcriptome analysis unveils novel pathway involved in the formation of yellow peel in cucumber. International Journal of Molecular Sciences, 22(3), 1494. https://doi.org/10.3390/ijms22031494 [Google Scholar] [PubMed] [CrossRef]
22. Fu, M., Yang, X., Zheng, J., Wang, L., Yang, X. et al. (2021). Unraveling the regulatory mechanism of color diversity in Camellia japonica petals by integrative transcriptome and metabolome analysis. Frontiers in Plant Science, 12, 685136. https://doi.org/10.3389/fpls.2021.685136 [Google Scholar] [PubMed] [CrossRef]
23. Wang, R., Ren, C., Dong, S., Chen, C., Xian, B. et al. (2021). Integrated metabolomics and transcriptome analysis of flavonoid biosynthesis in safflower (Carthamus tinctorius L.) with different colors. Frontiers in Plant Science, 12, 712038. https://doi.org/10.3389/fpls.2021.712038 [Google Scholar] [PubMed] [CrossRef]
24. Xiao, L., Cao, S., Shang, X., Xie, X., Zeng, W. et al. (2021). Metabolomic and transcriptomic profiling reveals distinct nutritional properties of cassavas with different flesh colors. Food Chemistry Molecular Sciences, 2, 100016. https://doi.org/10.1016/j.fochms.2021.100016 [Google Scholar] [PubMed] [CrossRef]
25. Yang, W., Su, Y., Dong, G. Q., Qian, G. T., Shi, Y. H. et al. (2020). Liquid chromatography-mass spectrometry-based metabolomics analysis of flavonoids and anthraquinones in Fagopyrum tataricum L. Gaertn (tartary buckwheat) seeds to trace morphological variations. Food Chemistry, 331(4), 127354. https://doi.org/10.1016/j.foodchem.2020.127354 [Google Scholar] [PubMed] [CrossRef]
26. He, M., He, Y. Q., Zhang, K. X., Lu, X., Zhang, X. M. et al. (2022). Comparison of buckwheat genomes reveals the genetic basis of metabolomic divergence and ecotype differentiation. New Phytologist, 235(5), 1927–1943. https://doi.org/10.1111/nph.18306 [Google Scholar] [PubMed] [CrossRef]
27. Shi, X. D., Wang, C., Li, W. J., Xiao, H. L., Li, Y. J. et al. (2022). Analysis of seed phenotypic and metabolic characteristics of diploid and tetraploid tartary buckwheat. Phyton-International Journal of Experimental Botany, 91(9), 1973–1986. https://doi.org/10.32604/phyton.2022.020065 [Google Scholar] [PubMed] [CrossRef]
28. Feldman, M., Levy, A. A. (2005). Allopolyploidy-a shaping force in the evolution of wheat genomes. Cytogenetic and Genome Research, 109(1–3), 250–258. https://doi.org/10.1159/000082407 [Google Scholar] [PubMed] [CrossRef]
29. Sattler, M. C., Carvalho, C. R., Clarindo, W. R. (2016). The polyploidy and its key role in plant breeding. Planta, 243(2), 281–296. https://doi.org/10.1007/s00425-015-2450-x [Google Scholar] [PubMed] [CrossRef]
30. Vande, P. Y., Mizrachi, E., Marchal, K. (2017). The evolutionary significance of polyploidy. Nature Reviews Genetics, 18(7), 411–424. https://doi.org/10.1038/nrg.2017.26 [Google Scholar] [PubMed] [CrossRef]
31. Osborn, T. C., Pires, J. C., Birchler, J. A., Auger, D. L., Chen, Z. J. et al. (2003). Understanding mechanisms of novel gene expression in polyploids. Trends in Genetics, 19(3), 141–147. https://doi.org/10.1016/S0168-9525(03)00015-5 [Google Scholar] [PubMed] [CrossRef]
32. Xu, C., Bai, Y., Lin, X., Zhao, N., Hu, L. et al. (2014). Genome-wide disruption of gene expression in allopolyploids but not hybrids of rice subspecies. Molecular Biology and Evolution, 31(5), 1066–1076. https://doi.org/10.1093/molbev/msu085 [Google Scholar] [PubMed] [CrossRef]
33. Tsukaya, H. (2013). Does ploidy level directly control cell size? Counterevidence from Arabidopsis genetics. PLoS One, 8(12), e83729. https://doi.org/10.1371/journal.pone.0083729 [Google Scholar] [PubMed] [CrossRef]
34. Bhattarai, K., Kareem, A., Deng, Z. (2021). In vivo induction and characterization of polyploids in gerbera daisy. Scientia Horticulturae, 282(2), 110054. https://doi.org/10.1016/j.scienta.2021.110054 [Google Scholar] [CrossRef]
35. Yu, Z., Haberer, G., Matthes, M., Rattei, T., Mayer, K. F. et al. (2010). Impact of natural genetic variation on the transcriptome of autotetraploid Arabidopsis thaliana. Proceedings of the National Academy of Sciences of USA, 107(41), 17809–17814. https://doi.org/10.1073/pnas.1000852107 [Google Scholar] [PubMed] [CrossRef]
36. Braynen, J., Yang, Y., Wei, F., Cao, G. Q., Shi, G. Y. et al. (2017). Transcriptome analysis of floral buds deciphered an irregular course of meiosis in polyploid Brassica rapa. Frontiers in Plant Science, 8, 768. https://doi.org/10.3389/fpls.2017.00768 [Google Scholar] [PubMed] [CrossRef]
37. Li, H. Y., Lv, Q. Y., Liu, A. K., Wang, J. R., Sun, X. Q. et al. (2022). Comparative metabolomics study of tartary (Fagopyrum tataricum (L.). Gaertn) and common (Fagopyrum esculentum Moench) buckwheat seeds. Food Chemistry, 371(2), 131125. https://doi.org/10.1016/j.foodchem.2021.131125 [Google Scholar] [PubMed] [CrossRef]
38. Zhao, H., Liu, H. Z., Jin, J. J., Ma, X. Y., Li, K. L. (2022). Physiological and transcriptome analysis on diploid and polyploid Populus ussuriensis Kom. under salt stress. International Journal of Molecular Sciences, 23(14), 7529. https://doi.org/10.3390/ijms23147529 [Google Scholar] [PubMed] [CrossRef]
39. Wu, M., Ge, Y., Xu, C., Wang, J. (2020). Metabolome and transcriptome analysis of Hexaploid Solidago canadensis roots reveals its invasive capacity related to polyploidy. Genes, 11(2), 187. https://doi.org/10.3390/genes11020187 [Google Scholar] [PubMed] [CrossRef]
40. Zhang, Z., Tan, M., Zhang, Y., Jia, Y., Zhu, S. et al. (2021). Integrative analyses of targeted metabolome and transcriptome of Isatidis Radix autotetraploids highlighted key polyploidization-responsive regulators. BMC Genomics, 22(1), 670. https://doi.org/10.1186/s12864-021-07980-w [Google Scholar] [PubMed] [CrossRef]
41. Balao, F., Herrera, J., Talavera, S. (2011). Phenotypic consequences of polyploidy and genome size at the microevolutionary scale: A multivariate morphological approach. New Phytologist, 192(1), 256–265. https://doi.org/10.1111/j.1469-8137.2011.03787.x [Google Scholar] [PubMed] [CrossRef]
42. Bertrand, B., Bardil, A., Baraille, H., Dussert, S., Doulbeau, S. et al. (2015). The greater phenotypic homeostasis of the allopolyploid coffea arabica improved the transcriptional homeostasis over that of both diploid parents. Plant and Cell Physiology, 56(10), 2035–2051. https://doi.org/10.1093/pcp/pcv117 [Google Scholar] [PubMed] [CrossRef]
43. Tamayo, M. C., Rodriguez, L. C., Narvaez, J. A., Tamayo, Y. J., Ayil, B. A. et al. (2016). Morphological features of different polyploids for adaptation and molecular characterization of CC-NBS-LRR and LEA gene families in Agave L. Journal of Plant Physiology, 195, 80–94. https://doi.org/10.1016/j.jplph.2016.03.009 [Google Scholar] [PubMed] [CrossRef]
44. Zhu, H. J., Liu, W. G. (2018). Progress on salt resistance in autopolyploid plants. Hereditas, 40(4), 315–326. https://doi.org/10.16288/j.yczz.17-305 [Google Scholar] [PubMed] [CrossRef]
45. Dai, F., Wang, Z., Luo, G., Tang, C. (2015). Phenotypic and transcriptomic analyses of autotetraploid and diploid mulberry (Morus alba L.). International Journal of Molecular Sciences, 16(9), 22938–22956. https://doi.org/10.3390/ijms160922938 [Google Scholar] [PubMed] [CrossRef]
46. Liao, T., Cheng, S., Zhu, X., Min, Y., Kang, X. (2016). Effects of triploid status on growth, photosynthesis, and leaf area in Populus. Trees-Structure and Function, 30(4), 1137–1147. https://doi.org/10.1007/s00468-016-1352-2 [Google Scholar] [CrossRef]
47. Allario, T., Brumos, J., Colmenero, J. M., Iglesias, D. J., Pina, J. A. et al. (2013). Tetraploid Rangpur lime rootstock increases drought tolerance via enhanced constitutive root abscisic acid production. Plant Cell and Environment, 36(4), 856–868. https://doi.org/10.1111/pce.12021 [Google Scholar] [PubMed] [CrossRef]
48. Saminathan, T., Nimmakayala, P., Manohar, S., Malkaram, S., Almeida, A. et al. (2015). Differential gene expression and alternative splicing between diploid and tetraploid watermelon. Journal of Experimental Botany, 66(5), 1369–1385. https://doi.org/10.1093/jxb/eru486 [Google Scholar] [PubMed] [CrossRef]
49. Del, J. C., Ramirez, P. E. (2014). Deciphering the molecular bases for drought tolerance in Arabidopsis autotetraploids. Plant Cell and Environment, 37(12), 2722–2737. https://doi.org/10.1111/pce.12344 [Google Scholar] [PubMed] [CrossRef]
50. Chen, Z. J. (2007). Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annual Review of Plant Biology, 58(1), 377–406. https://doi.org/10.1146/annurev.arplant.58.032806.103835 [Google Scholar] [PubMed] [CrossRef]
51. You, Q., Yi, X., Zhang, K., Wang, C., Ma, X. et al. (2017). Genome-wide comparative analysis of H3K4me3 profiles between diploid and allotetraploid cotton to refine genome annotation. Scientific Reports, 7(1), 9098. https://doi.org/10.1038/s41598-017-09680-6 [Google Scholar] [PubMed] [CrossRef]
52. Zhu, W., Hu, B., Becker, C., Dogan, E. S., Berendzen, K. W. et al. (2017). Altered chromatin compaction and histone methylation drive non-additive gene expression in an interspecific Arabidopsis hybrid. Genome Biology, 18(1), 157. https://doi.org/10.1186/s13059-017-1281-4 [Google Scholar] [PubMed] [CrossRef]
53. Ding, M. Q., Chen, Z. J. (2018). Epigenetic perspectives on the evolution and domestication of polyploid plant and crops. Current Opinion in Plant Biology, 42, 37–48. https://doi.org/10.1016/j.pbi.2018.02.003 [Google Scholar] [PubMed] [CrossRef]
54. Zou, L., Liu, W., Zhang, Z., Edwards, E. J., Gathunga, E. K. et al. (2020). Gene body demethylation increases expression and is associated with self-pruning during grape genome duplication. Horticulture Research, 7(1), 84. https://doi.org/10.1038/s41438-020-0303-7 [Google Scholar] [PubMed] [CrossRef]
55. Comai, L. (2005). The advantages and disadvantages of being polyploid. Nature Reviews Genetics, 6(11), 836–846. https://doi.org/10.1038/nrg1711 [Google Scholar] [PubMed] [CrossRef]
56. Madani, H., Escrich, A., Hosseini, B., Sanchez, R., Khojasteh, A. et al. (2021). Effect of polyploidy induction on natural metabolite production in medicinal plants. Biomolecules, 11(6), 899. https://doi.org/10.3390/biom11060899 [Google Scholar] [PubMed] [CrossRef]
57. Ravandi, E. G., Rezanejad, F., Dehghan, E. (2014). In vitro regeneration ability of diploid and autotetraploid plants of Cichorium intybus L. Cytology and Genetics, 48(3), 166–170. https://doi.org/10.3103/S0095452714030050 [Google Scholar] [CrossRef]
58. Lin, X., Zhou, Y., Zhang, J., Lu, X., Zhang, F. et al. (2011). Enhancement of artemisinin content in tetraploid Artemisia annua plants by modulating the expression of genes in artemisinin biosynthetic pathway. Biotechnology and Applied Biochemistry, 58(1), 50–57. https://doi.org/10.1002/(ISSN)1470-8744 [Google Scholar] [CrossRef]
59. Xu, Z., Gao, R., Pu, X., Xu, R., Wang, J. et al. (2020). Comparative genome analysis of Scutellaria baicalensis and Scutellaria barbata reveals the evolution of active flavonoid biosynthesis. Genomics Proteomics and Bioinformatics, 18(3), 230–240. https://doi.org/10.1016/j.gpb.2020.06.002 [Google Scholar] [PubMed] [CrossRef]
Supplementary Materials
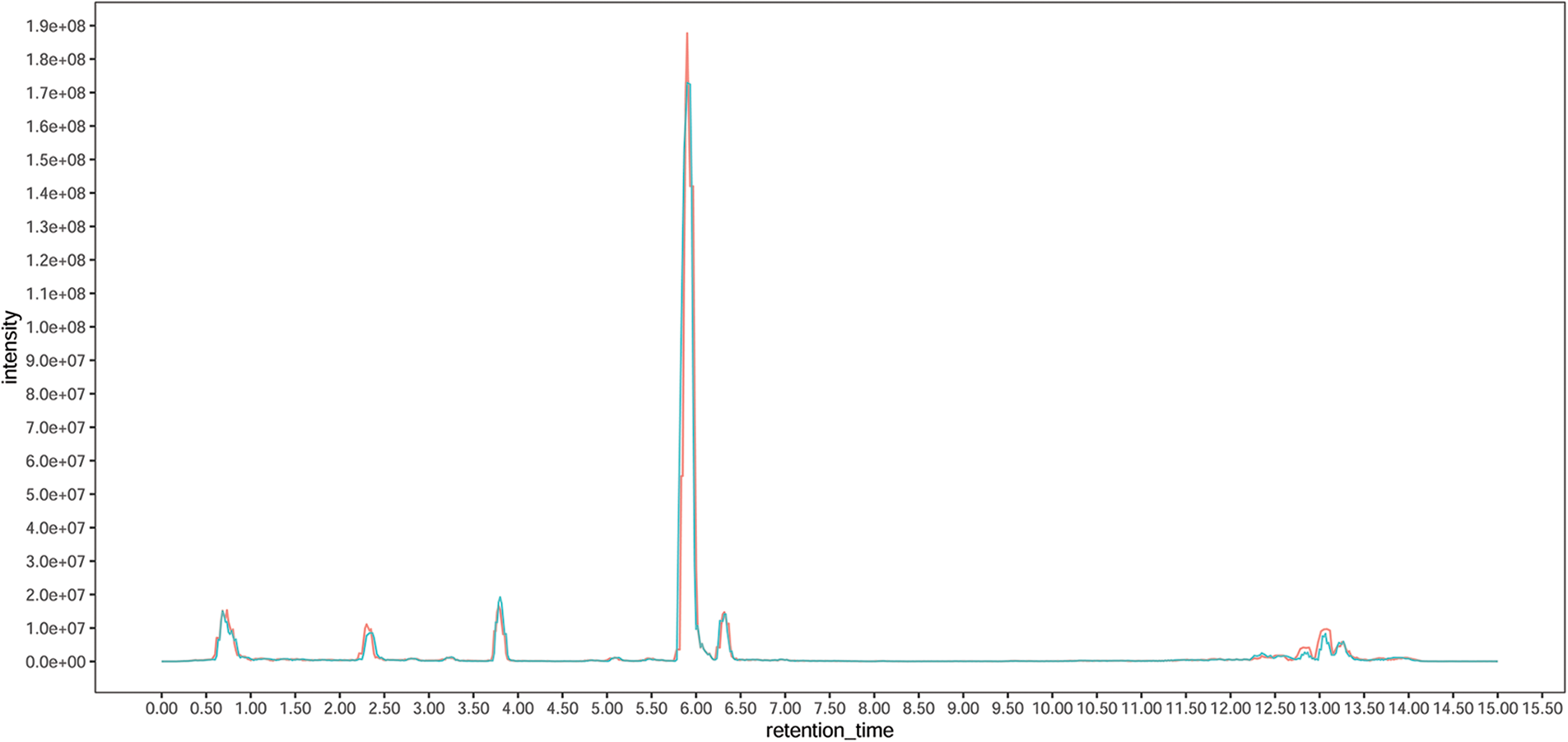
Supplementary Figure S1: The total ion current chromatograms of the different quality-control samples
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools