
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Multi-Omics Integration for Abiotic Stress Acclimation in Tropical and Underutilized Plants
1 Department of Plant Pathology, University of Agriculture, Faisalabad, 38000, Pakistan
2 Institute of Systems Biology (INBIOSIS), Universiti Kebangsaan Malaysia, UKM, Bangi, 43600, Selangor, Malaysia
* Corresponding Author: Hoe-Han Goh. Email:
(This article belongs to the Special Issue: Multi-Omics Insights into Plant Acclimation to Environmental Stress)
Phyton-International Journal of Experimental Botany 2026, 95(6), 3 https://doi.org/10.32604/phyton.2026.084657
Received 27 April 2026; Accepted 04 June 2026; Issue published 29 June 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Abiotic stresses particularly drought, salinity, and temperature extremes increasingly threaten crop stability in tropical environments where combinatorial stress events are intensifying under climate change. Despite growing omics capacity, current multi-omics syntheses of plant stress acclimation remain dominated by model species and major crops, leaving tropical and underutilized plants underrepresented in comparative stress biology. This review examines how transcriptomics, proteomics, metabolomics, epigenomics, and integrative multi-omics frameworks have been applied to abiotic stress acclimation in selected tropical and underutilized plants, including banana, cassava, cacao, oil palm, papaya, Garcinia, coconut, mango, Avicennia marina, and tropical millets. We show that transcriptomics has provided the foundation for stress-response characterization in these species, but that proteomics and metabolomics reveal acclimation processes including protein-level buffering, osmolyte accumulation, and specialized-metabolite remodeling not captured by transcript profiles alone. Multi-omics integration strengthens candidate prioritization by identifying molecular convergence across layers, though most regulatory candidates in tropical systems remain associative and require functional validation. Conserved acclimation mechanisms include ABA-responsive transcription factor induction, ROS-scavenging remodeling, heat shock protein accumulation, and osmolyte biosynthesis, while lineage-specific responses highlight additional biochemical diversity unique to tropical species. Key bottleneck including complete reference genomes, limited metabolite annotation, inconsistent phenotyping, and poor cross-study comparability continue to constrain mechanistic interpretation. Priority future directions include pangenome resource development, single-cell and spatial profiling, machine-learning-assisted integration, and validation-focused experimental design. By placing tropical and underutilized plants more centrally within plant stress biology, this review advocates a broader, more inclusive framework for understanding acclimation under intensifying global change.Graphic Abstract
Keywords
Abiotic stresses particularly drought, salinity, heat, cold, nutrient imbalance, and waterlogging remain among the most persistent constraints on plant growth, productivity, and yield stability across agricultural systems [1,2,3]. Their importance has grown sharply under accelerating climate change, which is intensifying the frequency, duration, and co-occurrence of environmental stresses, and is expected to depress global yields of major staple crops by 10–25% before mid-century unless adaptive strategies are successfully deployed [4]. Crop losses attributable to drought and heat alone have more than tripled over the past five decades, with combinatorial stress events particularly drought plus heat, or waterlogging plus heat causing disproportionately large reductions that cannot be predicted from single-stress responses [5]. Before proceeding, it is important to clarify the terminological framework used in this review. Stress response refers to any immediate molecular, biochemical, or physiological reaction triggered by an abiotic stress stimulus, including changes in gene expression, protein abundance, or metabolite levels that occur within minutes to hours of stress onset. Acclimation denotes a reversible, phenotypically sustained adjustment that develops over a longer timescale and improves organismal performance under a given stress condition without permanent genetic change. Stress priming describes the phenomenon whereby a prior sub-lethal stress exposure enhances the speed, magnitude, or efficiency of the response to a subsequent stress event. Stress memory refers to the molecular and epigenetic mechanisms, including persistent histone modifications, DNA methylation changes, and small RNA accumulation, that underlie the retention of priming effects beyond the initial stress exposure, in some cases across cell generations or developmental stages. Long-term stress tolerance describes constitutively or adaptively fixed capacities that allow a species or genotype to maintain growth and reproduction under chronic stress conditions. These distinctions matter because not every stress-responsive transcript, protein, or metabolite is functionally related to acclimation many represent transient damage-response or signaling events whose downstream contribution to sustained performance remains unresolved. Throughout this review, these terms are used with these specific meanings, and claims about acclimation are restricted to evidence that goes beyond immediate stress-responsive changes. In crop science and plant molecular biology alike, the central issue is therefore no longer whether plants respond to stress, but how they coordinate molecular and biochemical adjustment, physiological protection under compound stress, and longer-term acclimation [6].
Over the past two decades, advances in high-throughput sequencing, mass spectrometry, and computational biology have transformed the study of plant stress responses [7,8,9]. Transcriptomics first expanded the scale at which stress-responsive genes could be identified, whereas proteomics, metabolomics, epigenomics, and integrative systems analysis later showed that stress acclimation cannot be understood from transcript-level data alone [10,11,12]. Transcript abundance, protein accumulation, metabolite turnover, and regulatory persistence often diverge substantially in both timing and magnitude, particularly under prolonged or combined abiotic stress [5,13,14]. Recent multifactorial-stress analyses in tomato, for example, demonstrate that transcriptomic, proteomic, and metabolomic layers respond with distinct kinetics and only partially overlapping sets of regulated entities, a pattern also observed in potato under heat, drought, and waterlogging. For this reason, multi-omics integration has become a core direction in contemporary plant stress biology, linking molecular profiling with systems-level interpretation [15].
The value of multi-omics approaches lies precisely in this capacity to move beyond description. Single-omics studies can identify lists of responsive genes, proteins, or metabolites, but they often provide limited insight into causal relationships, pathway coordination, or the regulatory architecture underlying acclimation [4,10,11]. By contrast, integrative analysis relates transcriptional reprogramming to protein-level buffering and metabolic adjustment [14,16], while also connecting these layers to hormonal control, chromatin dynamics, and stress memory [14,17]. Contemporary reviews have therefore emphasized that multi-omics is not merely an accumulation of datasets, but an analytical framework for linking molecular layers that are otherwise interpreted in isolation [15].
Despite this progress, literature remains highly uneven in its taxonomic and agricultural coverage [11,18,19]. Broad syntheses of plant abiotic stress biology are still dominated by model plants and major crops, particularly Arabidopsis thaliana, rice (Oryza sativa), maize (Zea mays), wheat (Triticum aestivum), and soybean (Glycine max) [1,6,7]. This is understandable since these systems benefit from mature genomic resources, standardized phenotyping pipelines, comprehensive annotation databases, and extensively characterized regulatory networks [20,21]. Yet it has narrowed the comparative base from which stress-acclimation mechanisms are generalized. In many recent reviews, tropical and underutilized plants appear only marginally, usually as isolated examples rather than as central systems for integrative synthesis.
That imbalance is increasingly difficult to justify. Tropical and underutilized plant species are not merely peripheral to plant science; many are directly relevant to food and nutritional security, diversified agriculture, resilient production systems, and region-specific adaptation to environmental instability [18,19,22]. They also include species with distinctive metabolic profiles, unusual ecological tolerances, and underexplored regulatory biology [23]. Recent systematic review work has emphasized that underutilized crops deserve greater scientific attention because of their potential contribution to resilient and healthy agri-food systems, yet molecular studies on these species remain fragmented compared with those on major crops [24]. In practical terms, this means that potentially important insights into antioxidant defense, osmotic adjustment, metabolic plasticity, and stress-associated regulatory networks remain under-synthesized in precisely the species that may prove increasingly valuable under intensifying climatic stress.
This issue is especially important in tropical systems. Tropical fruit, medicinal, and underutilized plants often accumulate distinctive classes of specialized metabolites and show stress responses shaped by evolutionary histories different from those of temperate model crops [23,25]. Yet the omics literature on these taxa is frequently scattered across case studies, generated under non-comparable experimental conditions, or limited to a single molecular layer [11,18,24]. Incomplete genomes and weak annotation reduce functional interpretation [20,21,26], while poor cross-study comparability and limited follow-up validation further slow synthesis [27]. A dedicated review is therefore needed not because omics data on tropical and underutilized plants do not exist, but because these data have not yet been integrated into a coherent framework for understanding abiotic stress acclimation [10,19].
The present review addresses that gap by examining how transcriptomics, proteomics, metabolomics, epigenomics, and integrative multi-omics approaches have been applied to abiotic stress acclimation in selected tropical and underutilized plants. Rather than providing another broad summary of plant stress omics, the review focuses specifically on species and systems that remain comparatively underrepresented in mainstream syntheses [18,19,24]. It evaluates what multi-omics approaches have already clarified in these plants; where interpretation remains limited by methodological and resource constraints; and how future work can move from descriptive response profiling toward stronger biological inference [6,7,16] and translational payoff [14,20]. By situating tropical and underutilized species more centrally within the stress-omics literature, the review aims to broaden the conceptual base of plant stress biology and highlight a more inclusive framework for understanding acclimation in a changing environment [10].
This review was conducted as a narrative synthesis of the primary and review literature on multi-omics approaches to abiotic stress acclimation in tropical and underutilized plants. Literature was retrieved through systematic searches of Web of Science, Scopus, PubMed, and Google Scholar, using keyword combinations including “multi-omics”, “abiotic stress”, “tropical plants”, “underutilized crops”, “transcriptomics”, “proteomics”, “metabolomics”, “epigenomics”, “stress acclimation”, and related species-specific terms. The primary search timeframe was 2015–2026, with foundational earlier studies included where necessary to establish methodological or conceptual context. Species were selected on the basis of two criteria: “tropical” refers to species whose cultivation range falls predominantly within the Köppen tropical climate zones (Af, Am, Aw/As), and “underutilized” follows the Bioversity International definition crops with recognized nutritional, economic, or ecological value that remain underrepresented in mainstream scientific research relative to their potential. Coverage depth was intentionally uneven and reflects the actual state of the literature; species with richer multi-omics datasets receive more detailed treatment, while less-studied species are highlighted to indicate research gaps. This review is distinguished from existing multi-omics stress biology syntheses by its deliberate centering of tropical and underutilized species as the primary analytical frame rather than as peripheral examples within model-crop-focused narratives.
2 Tropical and Underutilized Plants Are Lacking in Mainstream Stress-Omics Synthesis
The underrepresentation of tropical and underutilized plants in mainstream stress-omics synthesis is not simply a consequence of scientific neglect at the level of individual species. Rather, it reflects a broader asymmetry in plant research infrastructure [20,21,28]. Major crops and model plants dominate omics-based stress biology because they benefit from a long-standing accumulation of genomic resources, curated annotations, standardized reference datasets, and analytical pipelines that make systems-level interpretation comparatively efficient [26,29]. Tropical and underutilized species, by contrast, often enter the literature only through isolated case studies, and their datasets are frequently insufficiently connected to broader comparative frameworks [19,24].
A major part of this imbalance lies in genome-resource availability [20,21,26]. Functional omics interpretation depends heavily on the quality of the underlying genome reference. In species lacking well-assembled and well-annotated genomes, transcriptomic profiling becomes more difficult to interpret, proteomic peptide assignment becomes less complete, and cross-layer integration becomes inherently less reliable [29]. Recent reviews on plant pangenomes and orphan crop genomics have emphasized that genomic underrepresentation is not merely a technical inconvenience; it is a limiting factor that shapes which species can meaningfully participate in comparative biology, trait dissection, and translational crop improvement [30]. As a result, many tropical and underutilized plants remain visible only at the level of fragmented discovery rather than full functional interpretation [18,24].
This resource disparity also reinforces publication and funding bias. Once a species becomes genomically tractable, it is more likely to attract further studies, higher-quality functional analyses, and broader review inclusion [18,26,28]. Species lacking such infrastructure remain trapped in a low-visibility cycle: fewer genomic tools lead to fewer mechanistic studies, which in turn lead to less synthesis and less future investment [19,24]. Recent studies on orphan and underutilized crops have highlighted this structural problem repeatedly, while broader crop-diversification and pangenome perspectives argue that resilience and nutrition security will depend in part on widening the crop research base [30,31].
Analytical constraints further amplify this imbalance. Multi-omics research in non-model plants is not limited by sequencing alone. Annotation quality, orthology inference, metabolite identification, and data harmonization all become more difficult in species with incomplete references or sparse prior literature [21,26,32]. Metabolomics is particularly affected because many tropical and underutilized plants produce lineage-specific or poorly catalogued compounds that remain weakly represented in standard spectral libraries [22,33,34]. Similarly, transcriptomic and proteomic datasets generated from different tissues, developmental stages, and stress regimes are difficult to compare when common reference frameworks are lacking [20,24,29]. This means that even when omics studies are available, they often remain descriptive rather than mechanistically integrative [18].
Language and geography reinforce these technical barriers. Tropical crop research is concentrated in institutions across South and Southeast Asia, Sub-Saharan Africa, and Latin America, where access to large sequencing centres, proprietary metabolomics platforms, and long-term computational infrastructure is often more limited than in temperate-country institutions [19,24,28]. Much of the resulting work is published in regional journals or specialty outlets that are less frequently screened by global systematic reviews, further compounding invisibility [18]. Addressing this imbalance therefore requires not only better tools but also deliberate curatorial effort to incorporate regional literature into comparative synthesis.
For these reasons, the absence of tropical and underutilized plants from mainstream stress-omics synthesis should not be understood simply as a gap in coverage; it reflects a deeper imbalance in how plant functional genomics has been organized [18,24,28]. A review focused specifically on abiotic stress acclimation in these species is therefore justified not only as a matter of representation, but as a necessary step toward broadening the comparative base of plant stress biology. Bringing these systems into clearer analytical focus may reveal conserved stress-acclimation mechanisms [19], as well as lineage-specific strategies that remain obscured when synthesis is restricted to a small set of well-resourced crops [30,31].
3 Transcriptomic Responses to Abiotic Stress in Tropical Species
Transcriptomics has been the most widely adopted omics approach in tropical and underutilized plant stress biology, largely because RNA-seq can be deployed even in species where proteomic, metabolomic, and genome-scale functional resources remain limited [6,10,16]. In many tropical species, transcriptomic profiling has provided the first system-wide view of abiotic stress responses, identifying candidate transcription factors, signaling components, transporters, and metabolic genes associated with acclimation [3,9,35]. At the same time, the interpretive value of these datasets depends heavily on experimental design, reference quality [21,29], and whether transcript-level patterns are later integrated with other biological layers. Representative transcriptome-led examples are summarized in Table 1.
Table 1: Representative transcriptome-led abiotic stress studies in tropical and underutilized plants.
| Species | Stress | Experimental Scope and Quantitative Output | Main Acclimation Insight | Ref. |
|---|---|---|---|---|
| Banana (Musa spp.) | Drought | mRNA-Seq of tolerant ‘Saba’ and sensitive ‘Grand Naine’; 162 million and 126 million reads, respectively; 2268 and 2963 differentially expressed genes (DEGs); transcription factors (MYB, WRKY, bHLH, NAC) accounted for ~15.9% of DEGs alongside kinases, lipid and carbohydrate remodeling, and secondary-metabolite genes. | Drought response was genotype-dependent, with strong representation of regulatory genes rather than a single conserved expression program. | [36] |
| Cacao (Theobroma cacao) | Drought | Integrated physiological measurements with RNA-Seq of two drought-tolerant hybrid clones. | Two tolerant clones reached drought tolerance through alternative molecular routes one emphasizing stomatal/photoprotection, the other osmotic/antioxidant adjustment. | [37] |
| Cassava (Manihot esculenta) | High temperature | Comparative leaf vs. mid-vein transcriptomics; 89.17 Gb clean reads; tissue-specific DEG sets for mesophyll and vasculature. | Leaves preferentially enhanced pyruvate synthesis, whereas mid-vein tissues remodeled sucrose/starch metabolism and phloem-associated transporters. | [38] |
| Oil palm (Elaeis guineensis) | Drought, salinity, waterlogging, heat, cold | Leaf RNA-Seq across five stresses; 19,834 total DEGs; 588 core DEGs shared across all five conditions. | A core abiotic stress transcriptome was identified, including kinases, NAC/ERF TFs, HSPs, ubiquitin components, and osmolyte/redox genes, alongside stress-specific DEG sets. | [39] |
Among tropical crops, drought remains the most extensively studied abiotic stress at the transcriptomic level. Early RNA-seq work in banana showed that drought-tolerant and drought-sensitive cultivars differed not only in the magnitude of their transcriptional response but also in the distribution of regulatory genes induced under water deficit [36]. In a comparative study of the tolerant cultivar ‘Saba’ and the sensitive cultivar ‘Grand Naine’, drought treatment altered the expression of 2268 and 2963 genes, respectively, with transcription factor families especially MYB, WRKY, bHLH, and NAC members accounting for approximately 15.9% of differentially expressed genes (DEGs) alongside protein kinases, lipid metabolism genes, carbohydrate remodeling enzymes, and secondary-metabolite-related pathways. That study remains important because it demonstrated that even within a single tropical crop, drought-responsive transcriptional reprogramming is genotype-dependent, suggesting that acclimation strategies may differ between cultivars, though functional confirmation of these differences remains limited.
More recent work has extended this picture by moving from static transcript catalogues toward more targeted regulatory interpretation. A study in Musa acuminata used time-course transcriptomics and downstream molecular analyses to identify MaGME777 and MabHLH770 as candidate regulators associated with drought-responsive expression, though their causal roles in tolerance remain to be validated functionally MaGME777 MabHLH770, illustrating the field’s gradual shift from transcript discovery toward candidate-gene resolution [40]. Complementary work on banana aquaporins has identified MaPIP11 as a stress-responsive water-transport component whose expression is associated with drought and salinity conditions, suggesting a potential role in acclimation, further refining the regulatory vocabulary of dehydration acclimation in Musa [41]. Although this remains far from a full multi-omics framework, it shows how transcriptomics in tropical crops is now beginning to generate more targeted candidate nominations rather than only descriptive DEG lists, even if functional validation of most candidates remains outstanding.
Cacao (Theobroma cacao) provides another strong example of the increasing maturity of transcriptomic stress research in tropical species. A 2026 integrated physiological and transcriptomic study of drought response in two cacao hybrid clones showed that drought tolerance may be achieved through distinct molecular routes rather than through a single common expression pattern: one clone relied predominantly on stomatal regulation and photoprotection, whereas a second achieved equivalent tolerance through enhanced osmotic adjustment and antioxidant buffering [37]. This is important for review framing because it emphasizes that transcriptomic variation among tolerant tropical genotypes may reflect alternative acclimation strategies, not merely stronger or weaker activation of the same pathways.
While drought has received the greatest attention, transcriptomic studies of other abiotic stresses are also increasing in tropical systems. In cassava (Manihot esculenta), a tropical staple crop of major importance in climate-vulnerable regions, heat-stress transcriptomics identified marked tissue-dependent differences in gene expression patterns. A 2023 study showed that heat-responsive molecular programs differed substantially between vasculature-associated tissues and leaf mesophyll: leaves preferentially enhanced pyruvate synthesis while mid-vein tissues showed stronger remodeling of sucrose and starch metabolism, carbon export, and phloem-associated transporters [38]. This observation is especially relevant in non-model tropical crops because many stress studies still rely on pooled tissue strategies that can obscure biologically meaningful cell- or tissue-level differences; emerging single-cell and spatial multi-omics approaches reinforce this point by showing how averaged tissue profiles can mask localized regulation [42] Complementary work has identified cassava A20AN1 zinc-finger proteins as candidates that interact with abiotic-stress signaling modules and are associated with drought and cold responses, providing a testable regulatory handle for future functional validation [43].
Oil palm (Elaeis guineensis) provides a useful example of transcriptomics at a broader stress-spectrum scale. A 2024 RNA-seq study examined leaf responses under drought, salinity, waterlogging, heat, and cold, and identified 19,834 DEGs in total across the five stresses, including 588 common DEGs shared across all five conditions [39]. The shared core set was dominated by kinases, NAC and ERF transcription factors, heat shock proteins, ubiquitin-related components, and genes associated with osmolyte biosynthesis, redox homeostasis, and secondary-metabolite pathways. The value of this work lies not only in the stress-specific DEG sets, but in the identification of a core set of stress-responsive transcripts shared across conditions, which provides a preliminary comparative framework for exploring how tropical crops may partition stress-general and stress-specific transcriptional responses. Similar comparative frameworks are beginning to emerge in other tropical crops, including date palm and pineapple, supporting the idea that stress-general regulatory backbones may be conserved across multiple tropical lineages [44].
Salinity and waterlogging studies in oil palm also highlight a broader point relevant to tropical plants: transcriptomic acclimation is often intertwined with environmental regimes that are especially important in humid tropical production systems, such as fluctuating rainfall, poor drainage, and combined osmotic stress [45]. A recent plantation-based transcriptomic study of oil palm stem responses to waterlogging showed that precipitation seasonality and field context shape expression outcomes in ways not easily reproduced in simplified controlled-environment experiments. This finding resonates with recent work in other tropical waterlogging-prone systems, including rice and banana, where root-oxygen availability and timing of flooding strongly influence transcriptomic outcomes [41,46]. It serves as a reminder that tropical stress transcriptomics often sit closer to ecological realism than many model-system studies but becomes correspondingly more difficult to standardize and compare.
Papaya (Carica papaya L.) represents another tropical fruit crop of direct relevance to abiotic stress transcriptomics, yet one that remains underexplored in proteomic and metabolomic terms. A tissue-resolved RNA-seq study of papaya under mild and severe drought—profiling leaves, sap, and roots across 18 cDNA libraries identified 8549 drought-responsive genes through reference-based analysis and 6089 drought-responsive unigenes through de novo assembly, with GO enrichment revealing that responses to ABA signaling, hormone signaling, sucrose metabolism, and suberin biosynthesis were strongly activated in roots under moderate drought stress, while oxidation-reduction and abiotic stress response pathways were enriched across all tissues under severe drought [47]. Co-expression network analysis of the same dataset identified 17 stress-related transcription factors as main regulatory hubs in leaves and roots, including WRKY70, MYB94, bHLH-ICE1, ABI5, ANAC072, and bZIP1 homologues, organized into tissue-specific and stress-severity-specific co-expression communities [47]. A complementary comparative transcriptomic study of a drought-tolerant wild papaya genotype against a susceptible commercial cultivar confirmed that six transcription factor genes CpHSF, CpMYB, CpNAC, CpNFY-A, CpERF, and CpWRKY were consistently more highly expressed in the tolerant genotype after 14 days of water deficit stress, with RT-qPCR validation confirming up to 47-fold higher expression levels for CpWRKY and 39-fold higher levels for CpHSF and CpMYB in the tolerant versus susceptible genotype [48]. Despite this growing transcriptomic foundation, proteomic and metabolomic characterization of Carica papaya stress responses is almost entirely absent from the current literature, positioning this species as a high-priority target for multi-omics integration among tropical underutilized fruit crops.
Underutilized cereals such as finger millet (Eleusine coracana), pearl millet (Cenchrus americanus), foxtail millet (Setaria italica), and proso millet (Panicum miliaceum) have likewise emerged as an important domain for tropical stress transcriptomics, combining intrinsic tolerance to drought, heat, and marginal soils with increasingly mature genome and transcriptome resources [49,50,51]. Transcriptomic studies in pearl millet and foxtail millet have highlighted drought- and heat-responsive networks involving hormone signaling, redox regulation, photosynthetic adjustment, and osmotic protection, reinforcing the view that millets provide tractable comparative systems for abiotic-stress biology. These transcriptomic foundations are revisited in a deeper case-study treatment in Section 7.5, where the broader multi-omics landscape in millets is considered alongside proteomic and metabolomic evidence.
Taken together, these studies show that transcriptomics has already generated substantial insight into abiotic stress acclimation in tropical species. Across crops such as banana [36,40,41], cassava [38,43], cacao [37], oil palm [39,45], and tropical millets [50,52,53], several recurring patterns are evident: strong reprogramming of regulatory genes, repeated involvement of hormone-responsive and redox-associated pathways, induction of transcription factor families such as MYB, WRKY, NAC, bHLH, and ERF, and substantial modulation of carbohydrate and secondary-metabolite metabolism. These are not trivial findings. They demonstrate that transcriptomics has been essential for bringing tropical species into molecular stress biology and for identifying the regulatory vocabulary of acclimation in species that historically lacked deep genomic resources.
At the same time, transcriptomics alone remains insufficient for a fully mechanistic account of stress acclimation. Differential expression does not necessarily predict protein abundance, enzyme activity, metabolite accumulation, or long-term physiological consequence [4,10,13]. This limitation is especially important in tropical and underutilized plants, where reference annotations remain incomplete [21,29] and many transcriptomic studies still end at pathway enrichment and candidate-gene nomination. As recent multi-omics reviews have emphasized, transcript-level studies are most informative when they serve as one layer in a broader integrative framework rather than as stand-alone evidence of regulatory mechanism [11,15]. For this reason, transcriptomics should be viewed not as the endpoint of stress-acclimation analysis in tropical plants, but as the foundation on which proteomic, metabolomic, and regulatory-level interpretation must be built.
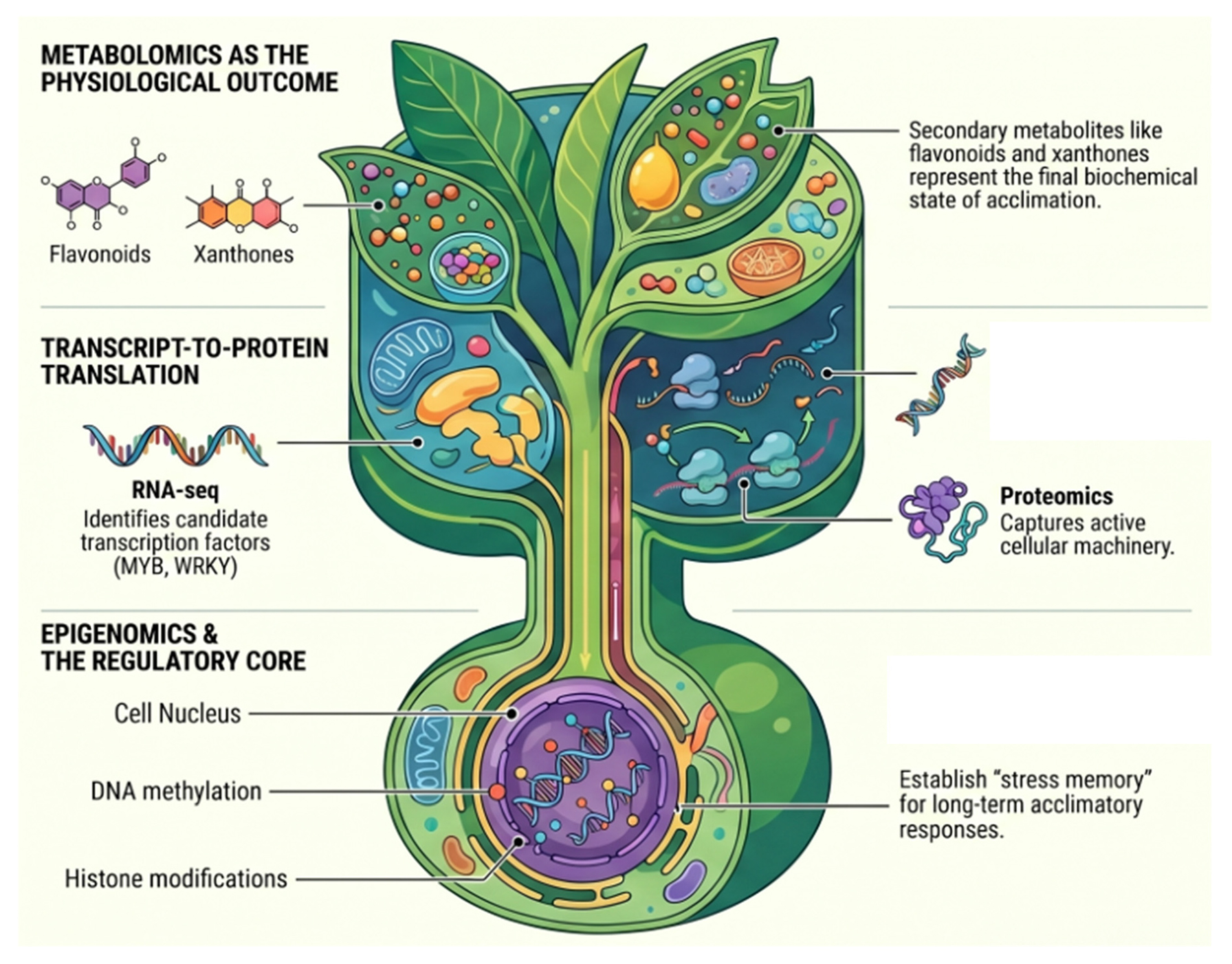
4 Proteomic and Metabolomic Perspectives on Stress Acclimation
Transcriptomics has provided the largest body of evidence for abiotic stress responses in tropical plants, but it captures only one layer of acclimation. Protein abundance and metabolite composition are more directly linked to stress physiology because they reflect the biochemical state of the cell at the point where regulation is enacted rather than only at the stage of transcript accumulation [13,33,54]. For this reason, proteomics and metabolomics are essential for moving beyond gene-level response catalogues toward a more functionally resolved view of acclimation. In tropical and underutilized plants, these two layers are especially important because many stress-related traits osmotic adjustment, antioxidant buffering, membrane stability, and secondary-metabolite remodeling are not adequately explained by RNA profiles alone [49,55].
Proteomic studies in tropical species remain fewer than transcriptomic studies, but they often provide stronger evidence of immediate functional adjustment under stress [54,55]. A representative example comes from banana, where iTRAQ-based proteomic analysis under salinity stress identified broad protein-level remodeling involving ROS-scavenging enzymes (SOD, APX, catalase), stress-related chaperones (HSP70, HSP90, sHSPs), photosynthetic machinery (RuBisCO small subunit, PSII proteins), ribosomal components, and proteins associated with energy metabolism [56]. This kind of evidence is important because it shows that acclimation in tropical crops is not simply a matter of altered transcript abundance, but also of selective stabilization, turnover, and reallocation of proteins directly tied to cellular protection and metabolic continuity. Tissue-specific proteomic work in pearl millet under drought stress has shown that protein-level reprogramming captures protective processes that are not always evident from transcript profiles alone, reinforcing the idea that proteome-level data can detect acclimation processes invisible at the transcriptome level [57].
Recent synthesis work in oil palm further reinforces this point. A 2024 review of low-temperature stress in oil palm concluded that proteomics has helped reveal post-transcriptional and biochemical adjustments not fully captured by transcriptomics, particularly in pathways related to redox homeostasis, membrane stability, photosynthesis, and stress-defense proteins [58]. Although much of the tropical stress literature is still transcript-heavy, the oil palm case illustrates that even in resource-limited perennial crops, proteomic data are beginning to refine the interpretation of acclimation by identifying molecular responses more closely linked to phenotype. Complementary proteomic analyses in cassava under combined drought and heat have revealed selective stabilization of cytosolic glutathione S-transferases and late embryogenesis abundant (LEA) proteins, extending the list of candidate acclimation proteins for tropical staples [59].
Metabolomics contributes a different kind of insight. While transcriptomics identifies candidate regulatory programs and proteomics captures parts of the active molecular machinery, metabolomics records the biochemical outcome of stress adjustment [22,32,33]. This is particularly relevant in tropical and underutilized plants, many of which are characterized by distinctive primary and secondary metabolite repertoires [23]. Metabolomics is especially useful for understanding osmotic adjustment, antioxidant status, central carbon metabolism, amino acid remodeling, and stress-associated specialized metabolites under drought, salinity, temperature stress, and related oxidative conditions [55,60].
An example is mango (Mangifera indica). In a combined metabolomic-transcriptomic study of cold stress in three mango cultivars, more than 1323 metabolites were detected and 22,526 DEGs were identified; the tolerant genotype showed stronger accumulation of flavonoids, terpenoids, lignans, coumarins, alkaloids, and multiple amino acid-related compounds under prolonged cold exposure [61]. These metabolite shifts were accompanied by transcriptomic evidence for altered phytohormone signaling, MAPK-related responses, and ICE–CBF–COR-associated cold-response pathways, but the metabolomic data were critical in showing the biochemical direction of acclimation rather than only its regulatory potential. The study is especially useful for this review because it demonstrates how tropical fruit stress biology can be interpreted through coordinated changes in both primary and secondary metabolisms.
Another example comes from coconut (Cocos nucifera). An integrated transcriptomic and metabolomic study of cold-stressed coconut seedlings identified 9968 DEGs and marked changes in amino acids, flavonoids, lipids, and phenolic-related compounds, together with stress-linked pathway activation in carbon metabolism, hormone signaling, and oxidative defense [62]. Although the study itself is integrative, its metabolomic dimension is especially relevant here because it shows how tropical perennial species may rely on metabolite reconfiguration as a major component of cold acclimation, even when their transcriptomic responses alone would be difficult to interpret fully. In tropical crops where growth and productivity are tightly constrained by relatively narrow thermal windows, such biochemical data are essential for understanding why some genotypes or species show stronger acclimatory capacity than others. Parallel metabolomics work on underutilized melon genotypes has similarly highlighted genotype-dependent accumulation of organic acids, amino acids, and flavonoid glycosides as correlates of water-deficit tolerance [60].
Proteomics and metabolomics are particularly valuable in tropical and underutilized species because they partly compensate for the interpretive limitations of incomplete genome annotation. In species where transcriptomic datasets are available, but gene models remain uncertain, protein-level evidence can validate which stress-associated pathways are functionally deployed, while metabolomic profiling can reveal whether those pathways converge on meaningful physiological outcomes such as osmoprotection, redox buffering, or secondary-metabolite adjustment [21,32,58]. Recent methodological reviews therefore argue that metabolomics and proteomics should not be treated as optional add-ons to transcriptomics, but as essential components of serious plant stress-acclimation analysis [4,16,33].
At the same time, these two approaches present challenges in tropical plants. Protein extraction is often complicated by high levels of polysaccharides, polyphenols, and interfering secondary compounds, requiring optimized TCA–acetone or phenol-based protocols that are still not widely benchmarked across tropical species [54,58]. Metabolite annotation remains difficult when tropical species produce lineage-specific compounds absent from standard databases such as HMDB, METLIN, or MassBank, often leading to large fractions of ‘unknown’ features in untargeted datasets [32,33,60]. Recent advances in computational metabolomics, including MS2 spectral prediction, in silico fragmentation, and deep-learning-based structure elucidation tools such as SIRIUS and MS2DeepScore, are beginning to narrow this gap, but their validation in tropical species is still preliminary. These technical constraints explain why transcriptomics still dominates literature, but they do not reduce the importance of proteomic and metabolomic data. On the contrary, they underscore the need for stronger biochemical characterization in tropical systems if abiotic stress acclimation is to be interpreted mechanistically rather than only descriptively.
Taken together, current evidence shows that proteomics and metabolomics already add substantial interpretive depth to tropical plant stress biology. Proteomics clarifies which stress-responsive proteins are accumulated or remodeled [56,57,58], while metabolomics reveals the biochemical phenotype of acclimation across central metabolism, osmotic adjustment, antioxidant defense, and specialized metabolite pathways [33,60,61]. In non-model and underutilized tropical species, these approaches are still developing, but they already demonstrate why transcriptomics alone cannot serve as a sufficient proxy for stress acclimation [13]. This transition from isolated molecular layers to integrated biological inference, illustrated with species-specific examples from the present review, is summarized in Fig. 1.

Figure 1: From isolated omics layers to integrated biological inference in tropical and underutilized plant stress biology. Transcriptomics identifies stress-responsive expression programs and candidate regulatory networks (e.g., ABA-responsive MYB/NAC induction in banana; 588-gene core stress transcriptome across five stresses in oil palm; tissue-specific heat responses in cassava vasculature vs. mesophyll; drought-responsive genotype-dependent transcriptional programs in cacao). Proteomics resolves protein-level buffering and functional adjustment that frequently diverges from transcript-level patterns (e.g., iTRAQ-based ROS-scavenging enzyme and chaperone remodeling in banana under salinity; tissue-specific drought proteome in pearl millet revealing redox-buffering proteins absent from transcript profiles; LEA protein and glutathione S-transferase stabilization in cassava under combined stress; post-transcriptional adjustments in oil palm under low-temperature stress). Metabolomics records the biochemical outcome of acclimation, including osmolyte accumulation, antioxidant remodeling, and specialized-metabolite shifts (e.g., flavonoid and terpenoid accumulation under cold stress in mango). Stronger biological inference emerges when these layers are aligned at the integration hub through pathway convergence, regulatory coordination, physiological relevance, and multi-layer candidate prioritization.
5 Integrating Omics Layers: From Descriptive Profiles to Biological Inference
The central promise of multi-omics integration lies not in the accumulation of additional datasets, but in the transition from molecular description to biological interpretation [10,11,16]. In plant stress biology, transcriptomic studies have been highly effective in identifying responsive genes and candidate regulatory pathways, yet they rarely resolve whether those changes translate into altered protein abundance, biochemical adjustment, or meaningful physiological acclimation [13,15]. Multi-omics integration is therefore valuable because it allows evidence from different biological layers to be considered together, increasing confidence in candidate mechanisms and reducing the tendency to over-interpret single-omics patterns [63]. In tropical and underutilized species, where genome annotation is often incomplete and functional validation remains limited, this integrative logic becomes even more important [21,29].
A useful conceptual starting point is provided by the systematic multi-omics integration (MOI) framework proposed by Jamil and colleagues, which distinguishes between element-based, pathway-based, and mathematical integration strategies [4]. Element-based integration links features across layers through shared identifiers (for example, mapping transcripts to proteins to metabolites within a single pathway), pathway-based integration aligns layer-specific enrichment results, and mathematical integration applies dimensionality-reduction or network inference methods such as MOFA+, DIABLO, or multi-omics factor analysis to identify latent factors that span datasets [11,64]. This framework is particularly relevant to non-model plants because it recognizes that not all studies need to achieve full-scale data fusion to be biologically useful. Even partial integration such as connecting transcriptomic and metabolomic patterns at the pathway level, or interpreting transcriptomic data alongside physiological measurements can substantially strengthen inference when reference resources are limited [63]. In the context of tropical stress biology, this is an important point: mechanistic insight often emerges not from perfect data completeness, but from careful alignment of complementary evidence. Representative tropical case studies are summarized in Table 2.
Integrated studies in tropical species already illustrate this advantage. In coconut, combined transcriptomic and metabolomic analysis under cold stress identified coordinated changes in phytohormone signaling, amino acid metabolism, flavonoid biosynthesis, and lipid-associated pathways, producing a more interpretively complete picture of candidate stress responses than either layer could provide on its own [62]. The value of that study was not merely that it produced two datasets, but that metabolite shifts helped prioritize which transcriptomic changes were most plausibly connected to biochemically enacted stress responses under cold exposure. Similarly, in mango, integrated transcriptome–metabolome profiling across cultivars with different cold tolerance showed that differential accumulation of amino acids, flavonoids, terpenoids, and other compounds could be interpreted together with transcriptional changes in ICE–CBF–COR signaling, phytohormone responses, and MAPK-associated pathways [61]. In both cases, integration moved the analysis from what changed at the transcript level to which coordinated molecular processes are most plausibly associated with tolerance, though causal confirmation requires downstream functional validation.
Table 2: Representative recent multi-omics abiotic-stress studies in tropical plants (transcriptome, proteome, physiology and/or metabolome).
| Species | Stress | Integrated Layers and Quantitative Output | Key Integrated Interpretation | Ref. |
|---|---|---|---|---|
| Coconut (Cocos nucifera) | Cold | Physiology plus transcriptome and metabolome of cold-stressed seedlings; 9968 DEGs and multiple metabolite classes (amino acids, lipids, flavonoids, phenolics). | Integration linked transcriptional shifts with biochemical adjustments in carbon metabolism, hormone signaling, and oxidative defense, identifying cold-acclimation pathways. | [62] |
| Mango (Mangifera indica) | Cold | Combined metabolome and transcriptome of three cultivars after prolonged cold; 1323 metabolites and 22,526 DEGs detected. | The tolerant cultivar showed stronger accumulation of flavonoids, terpenoids, lignans, coumarins, and alkaloids, coordinated with ICE–CBF–COR and MAPK pathways. | [61] |
| Avicennia marina (mangrove) | Low vs. high salinity | Root ionomics, transcriptomics and metabolomics under low and high salinity. | Low salinity favored HAK8-mediated K+ uptake and ABA signaling; high salinity relied on Na+ sequestration, glycine betaine accumulation, and polyphenol remodeling. | [65] |
| Pearl millet (Cenchrus americanus) | Salinity | Comparative physiology plus proteomics of contrasting genotypes under 150 mM NaCl; stress-associated proteins were interpreted alongside Na+/K+ balance, antioxidants, and membrane integrity. | Salt tolerance was associated with osmotic and ionic homeostasis, strong antioxidant capacity, photosynthetic maintenance, and a poor correlation between protein abundance and transcript levels. | [66] |
In tropical crops, an important intermediate form of integration involves combining molecular profiling with physiological data. Although such designs are not always presented as full multi-omics studies, they often provide stronger biological interpretation than omics datasets alone [14,15,67]. For example, recent work in oil palm integrated transcriptomic profiling with physiological drought-response measurements and showed that molecular signatures can be interpreted more usefully when considered alongside growth, water status, and stress-associated functional traits, though this does not on its own resolve causal mechanisms [68]. Likewise, a 2026 cacao study demonstrated that two drought-tolerant hybrid clones could achieve tolerance through different combinations of physiological behavior and transcriptomic reprogramming, including osmoprotection, antioxidant regulation, photoprotection, and ABA-associated responses [37]. Integrated root-ionome, transcriptome, and metabolome analyses in the mangrove Avicennia marina (Table 2) have similarly shown that low-salinity conditions were associated with HAK8-mediated K uptake and ABA signaling at the transcript and metabolite levels, whereas high-salinity conditions were associated with Na sequestration, glycine betaine accumulation, and polyphenol remodeling—a pattern consistent with tiered acclimation strategies, though direct functional validation of these pathway assignments is still needed [65]. These studies are especially relevant to the present review because they show that in tropical perennial and halophytic species, acclimation cannot be reduced to differential expression alone; interpretation becomes much stronger when gene-level changes are considered together with plant-level performance under stress.
This broader view of integration is important for tropical and underutilized plants because fully developed transcriptome–proteome-metabolome datasets are still relatively uncommon in many of these systems [18,24,28]. In practice, the most informative studies are often those that combine whichever molecular layer is currently available with well-designed physiological or biochemical datasets. From a review perspective, this means integration should not be defined too narrowly. If the goal is to explain stress acclimation rather than only to profile response, then physiological and phenotypic evidence must be considered part of the integrative framework, especially in species where deeper resource development is still in progress [37,65,68].
Multifactorial and combinatorial-stress studies offer a particularly strong argument for integration [2,5,13]. A 2025 analysis of potato responses to combined heat, drought, and waterlogging showed that only 9% of stress-regulated genes were shared across all three conditions, but that combined stress triggered a distinct multi-layer signature involving ABA–ethylene crosstalk, proline and trehalose accumulation, and remodeled lipid composition that could not have been predicted from single-stress datasets. Analogous tomato multifactorial studies have shown that integrated analysis of transcript, protein, metabolite, and ionomic data reveals emergent response modules absent from any single stress. These findings matter for tropical biology because combinatorial stresses, especially drought plus heat, waterlogging plus salinity, and heat plus cold-night transitions dominate many tropical production environments.
A second major contribution of integration is candidate prioritization. In non-model tropical species, stress studies often generate long lists of differentially expressed genes, abundant proteins, or responsive metabolites, but relatively few of these candidates can be followed up experimentally [4,11,63]. Multi-omics integration helps reduce this problem by identifying points of convergence across evidence layers. Candidates supported by transcript abundance, pathway placement, metabolite association, and, where possible, physiological correlation are more likely to be biologically meaningful than those emerging from a single dataset alone [64]. This is one of the most important reasons why integration is not simply technical refinement but a strategic necessity in resource-limited systems.
Recent tropical studies show how this works in practice. In coconut, integrated analyses narrowed broad stress-response datasets toward specific pathways and compounds likely to be involved in tolerance, including amino acid metabolism, flavonoid remodeling, and hormone-associated regulatory circuits [62]. Comparable integrative work in mango highlighted coordinated metabolite and transcript shifts linked to cold tolerance [61]. In oil palm, comparative transcriptome work across drought, salinity, waterlogging, heat, and cold identified a core abiotic-stress network that may provide a useful scaffold for interpreting pathway convergence across stress types [39]. In pearl millet, comparative physiological and proteomic analysis highlighted stay-green behavior, root morphological regulation, and photosynthetic protection as key features of drought resilience [52]. Together, these studies suggest that the future value of multi-omics in tropical plants lies less in producing ever larger stress-response inventories than in generating more selective and biologically justified candidate sets for downstream validation.
Despite these advances, the transition from descriptive profiles to biological inference remains uneven. Multi-omics integration in tropical plants is still constrained by small sample numbers, inconsistent stress protocols, incomplete annotations, low metabolite-identification confidence for lineage-specific compounds, and limited cross-study comparability [4,32,33]. These constraints are not trivial. They affect the quality of network inference, the reproducibility of pathway-level conclusions, and the confidence with which candidate regulators can be prioritized [63,64]. In many tropical studies, integration still occurs after separate layer-specific analyses rather than through deeper model-based frameworks. That limitation does not invalidate current work, but it does mean that the literature remains stronger at identifying stress-associated molecular signatures than at fully resolving causal regulatory architecture [11].
For this reason, the current value of integrative studies in tropical and underutilized plants lies in two related contributions. First, they improve interpretation within individual species by connecting molecular layers that would otherwise remain isolated [4,16]. Second, they provide a basis for more rigorous comparative synthesis, allowing tropical systems to be discussed in terms of coordinated stress biology rather than only as disconnected omics case studies [18,24,28]. As more such datasets accumulate, the field will be better positioned to distinguish conserved acclimation strategies from lineage-specific responses, which is ultimately the main comparative contribution that tropical and underutilized species can make to plant stress multi-omics [19,30,31]. This general workflow is summarized in Fig. 2.

Figure 2: Conceptual workflow for multi-omics integration in abiotic stress acclimation studies of tropical and underutilized plants. The diagram moves from different omics data layers through integration and biological inference to downstream candidate prioritization for validation.
6 Regulatory and Epigenetic Dimensions of Stress Acclimation
The transcriptome, proteome, and metabolome represent downstream layers of a broader regulatory system that determines how stress signals are perceived, interpreted, and translated into acclimatory responses [8,17,69]. In plants, these regulatory layers include transcription factor activity, RNA-based post-transcriptional control, chromatin-associated regulation, and various forms of stress memory [70,71,72]. Although these processes are now recognized as central to abiotic stress biology, their characterization remains highly uneven across the plant kingdom. In tropical and underutilized species, the regulatory dimension of stress acclimation is still less developed than the descriptive omics literature, with many datasets pointing toward likely regulators but relatively few studies progressing to mechanistic validation [35].
6.1 Transcription Factor Networks in Tropical Stress Biology
Transcription factors TFs are among the most frequently implicated regulatory nodes in transcriptomic studies of abiotic stress, where they repeatedly appear as candidates linking environmental perception to coordinated gene expression programs [35,70]. Across plant systems, families such as MYB, WRKY, NAC, DREB/CBF, bZIP, bHLH, and HSF repeatedly appear in drought, salinity, and temperature-stress pathways, often acting through combinatorial cis-regulatory logic rather than in isolation [73,74]. Recent synthesis has emphasized that these TF families remain central not only to the induction of stress-responsive genes, but also to the integration of hormonal signaling, reactive oxygen species homeostasis, and developmental trade-offs under stress. Contemporary review work counts over 60 distinct TF families implicated in plant abiotic stress, with combinatorial control being the rule rather than the exception.
In tropical and underutilized plants, however, TF biology is still much stronger at the level of candidate identification than at the level of functional validation. Many transcriptomic studies in banana [36], cacao [37], cassava [38], oil palm [39], mango [61], coconut [62], and pearl millet [49,51] have identified stress-responsive MYB, WRKY, NAC, ERF, and bHLH family members as candidates, but only a small number of these candidates have been evaluated through promoter assays, target-gene analysis, protein–protein interaction mapping, or direct functional testing. This means that the regulatory architecture of stress acclimation in tropical species is still often inferred from expression patterns rather than experimentally resolved. Even relatively modest regulatory studies are therefore highly informative because they establish a bridge from omics association to regulatory mechanism.
Mangosteen (Garcinia mangostana) offers a useful example. A MYB transcription factor regulating anthocyanin biosynthesis during mangosteen fruit ripening was identified experimentally as early as 2009, showing that MYB-based regulation of secondary metabolism is not merely inferred in this species but has direct molecular support [74]. More recently, the in silico characterization of mangosteen MYB transcription factors associated with anthocyanin and xanthone production has expanded the candidate regulatory framework and created a clearer basis for comparative follow-up [75]. Parallel WRKY-family analyses in tropical fruit systems such as passion fruit identify stress- and flavonoid-associated WRKY candidates with hormone- and light-responsive promoter elements, underscoring how broader TF resources can inform future comparative work in Garcinia [76]. Although these studies are developmental or bioinformatic rather than strictly stress-centered, they are directly relevant here because secondary-metabolite regulation, TF control, and environmental responsiveness often intersect in tropical fruit biology. Together, they illustrate how regulatory analysis in Garcinia can move from isolated gene-level observations toward a more comparative framework.
In rice, one OsSAP8 promoter was recently shown to respond to multiple abiotic stresses through cis-regulatory logic linked to stress and phytohormone responsiveness [77]. That work is not about a tropical underutilized species, but it remains useful here because it provides a concrete example of promoter-level abiotic stress regulation in a monocot staple that complements the bioinformatic regulatory frameworks emerging in Garcinia. It also highlights an important general point: promoter analysis and cis-regulatory dissection remain underused in tropical stress biology, even though they are crucial for moving from candidate-gene lists to mechanistic models of stress-responsive gene regulation [35].
6.2 RNA-Based Regulation and Stress Memory
RNA-based regulation has become increasingly important in contemporary models of plant stress memory and acclimation [69,78,79]. Small RNAs—particularly microRNAs (miRNAs), phased small interfering RNAs (phasiRNAs), and other regulatory RNA classes—can modulate transcription factor abundance, transcript stability, translational efficiency, and the persistence of stress-responsive states. Recent reviews have emphasized that RNA-based regulation is not simply an auxiliary layer, but a key component of how plants retain, attenuate, or reconfigure responses after an initial stress exposure. Additional layers including long non-coding RNAs (lncRNAs), circular RNAs (circRNAs), and alternative splicing further expand the regulatory repertoire [80,81].
In tropical and underutilized species, this dimension remains much less developed than transcriptomics or even metabolomics. A few studies in tropical crops have reported stress-responsive small RNAs or predicted miRNA targets in banana, cassava, and millet systems [69,81], but comprehensive regulatory interpretation is still uncommon. This is one reason current multi-omics research in tropical plants often remains stronger at the level of pathway association than at the level of post-transcriptional mechanism. From a review standpoint, this does not mean the field is empty; rather, it means that RNA-based regulation should be treated as one of the most important underdeveloped directions in tropical stress biology [78,79].
6.3 Epigenetic Regulation, Chromatin Dynamics, and Stress Memory
Epigenetic regulation adds another layer of complexity by influencing how prior stress exposure affects later responses. DNA methylation, histone post-translational modifications (PTMs), chromatin accessibility, nucleosome positioning, and RNA-directed DNA methylation (RdDM) can all contribute to what is often described as stress memory or priming [71,72,79]. A 2023 synthesis of histone PTMs under abiotic stress emphasized that stress-induced acetylation of H3K9 and H3K27 at drought- and heat-responsive loci, combined with demethylation of repressive H3K27me3 marks, provides a chromatin-level mechanism for priming-based memory in plants [82]. A 2024 review on heat-stress memory extended this picture by identifying HSFA2–HSP101–heat-stress granule modules and sustained H3K4me3 enrichment at memory genes as key features of somatic heat-stress recall [83]. Recent reviews in this area have collectively stressed that abiotic stress acclimation should not be viewed only as an immediate response, but also as a process shaped by regulatory persistence across time and sometimes across generations [84].
Here again, the tropical and underutilized plant literature is still relatively sparse. Most detailed mechanistic work on epigenetic stress memory comes from model species, where chromatin profiling techniques such as ATAC-seq, ChIP-seq for histone PTMs, whole-genome bisulfite sequencing (WGBS), and Cut&Run profiling are routinely applied to characterize priming loci [72,82,83]. Tropical species are more often represented by broad transcriptomic or metabolomic studies without parallel epigenetic analysis. This creates a major gap in interpretation: stress-associated expression changes may be visible, but the persistence, reversibility, and regulatory memory of those changes remain poorly resolved. Preliminary DNA methylome studies in cassava and rubber tree suggest that drought-responsive differentially methylated regions (DMRs) concentrate near stress-associated TFs and transposable elements, but systematic transgenerational analyses are still lacking [84]. As a result, epigenetic stress memory remains one of the most conceptually important yet empirically weakest areas in current tropical stress multi-omics [71,79].
The relative weakness of regulatory and epigenetic studies in tropical and underutilized plants is not surprising. These analyses generally require better genome assemblies and stronger promoter annotation [21,26], more precise time-course and repeated-stress experimental frameworks [72,82], and more demanding experimental validation than transcript-level studies [83]. In species where even baseline genome resources are recent or incomplete, it is much easier to generate lists of differentially expressed genes than to test regulatory interactions directly. Yet the field cannot remain indefinitely at the descriptive stage. If abiotic stress acclimation in tropical systems is to be interpreted mechanistically, future studies will need to move more consistently toward TF validation and cis-regulatory analysis [35], RNA-based control [69], and stress-memory frameworks [71,79].
For this reason, the regulatory and epigenetic dimension should be viewed not as an optional extension to tropical stress omics, but as one of its major future priorities. Current omics studies have already identified many plausible regulatory candidates. The next stage is to determine which of these candidates actually coordinate stress acclimation, how their downstream targets are organized, and whether their effects remain transient or become part of a longer-term acclimatory memory. Integrating TF validation and RNA-based regulation [35,69] with chromatin and stress-memory analysis [71,72,83] will be essential if tropical multi-omics is to deliver more than descriptive snapshots of stress response.
7 Case Studies from Tropical and Underutilized Plants
Case studies are most useful in this review when they do more than illustrate species diversity. They should also show how far stress multi-omics has progressed in different tropical and underutilized systems, and where the main bottlenecks remain [18,24,28]. The examples selected here therefore represent different stages of analytical maturity: resource-enriched but stress-underdeveloped fruit systems such as Garcinia; tropical staple crops moving from transcriptomics toward broader systems-level integration such as cassava; fruit crops with recent integrative cold-stress datasets such as coconut and mango; ecologically specialized halophytic systems in which multi-omics has begun to illuminate salinity acclimation directly, as in Avicennia marina; and underutilized cereals such as tropical millets. Together, these cases show that tropical plant research is moving beyond data absence [19], but still faces the challenge of converting fragmented datasets into coherent biological inference [30,31].
7.1 Garcinia mangostana: From Resource Development to Stress Biology
Garcinia mangostana is highlighted in this review not as an established model for abiotic stress biology, but as a resource-ready yet stress-underexplored sytem. Its existing transcriptomic, proteomic, metabolomic, and regulatory infrastructure makes it a strong candidate for future stress multi-omics investigation, even though several of the studies cited in this section address developmental regulation and secondary metabolism rather than direct abiotic stress responses.
Among underutilized tropical fruit crops, Garcinia mangostana is one of the clearest examples of a system in which resource development has moved ahead of mechanistic stress biology. Over the last few years, mangosteen research has gained a stronger omics foundation through the development of a genome resource [85], organelle-genome studies of the mitochondrion [86] and plastome [87], a ripening transcriptome [88], an eFP reference expression atlas [89], and a recent candidate MYB TF framework associated with anthocyanin and xanthone biosynthesis [75]. SWATH-MS quantitative proteomics of ripening fruit pericarp has added the first protein-level layer to these resources, resolving stress-associated chaperones, ROS-scavenging enzymes, and secondary-metabolite biosynthetic proteins co-expressed with ripening-associated genes [90]. Complementing this, ESI-LC-MS untargeted metabolomics across pericarp, aril, and seed at successive ripening stages has catalogued xanthones, prenyl-flavonoids, and osmoprotective compounds enriched in specific tissues [91]. Together, these resources mean that Garcinia is no longer an omics-poor system in the same way many underutilized species still are. At the same time, direct multi-omics studies of abiotic stress acclimation in mangosteen remain scarce, so the species currently represents a platform with strong translational potential but limited stress-specific systems-level interpretation.
This makes Garcinia especially instructive for the present review. Its value lies not only in fruit quality and secondary-metabolite richness, but also in the way it exposes an important asymmetry in tropical plant research: developmental and metabolite-associated omics resources may be available, yet stress-acclimation biology can remain poorly integrated. Relevant Garcinia resources already span the MYB regulatory framework [75], a comparative tropical-fruit WRKY framework [76], the genome resource [85], mitochondrial and plastome references [86,87], the ripening transcriptome [88], the eFP atlas [89], and more recent proteome and metabolome studies [90,91]. Comparative extension into related taxa such as G. atroviridis is also attractive because these species add phytochemical and functional diversity relevant to nutraceutical and medicinal plant research, while still remaining anchored within a Southeast Asian tropical context [92]. In this sense, Garcinia illustrates both the progress and the current incompleteness of tropical fruit multi-omics. The available resources are summarized in Table 3.
Table 3: Published Garcinia resources and selected comparative regulatory studies relevant to future abiotic-stress integration.
| Resource/Study | Published Content | Strategic Value for Stress Multi-Omics | Ref. |
|---|---|---|---|
| Genome resource review | Dedicated genome chapter/resource for mangosteen | Shows that Garcinia is no longer a genomically invisible system; provides a reference base for comparative stress genomics. | [85] |
| Mitochondrial genome (var. Mesta) | Complete mitochondrial genome | Demonstrates organelle-level resource development; supports future studies of mitochondrial contribution to abiotic stress tolerance. | [86] |
| Plastome comparative analysis (G. mangostana and related species) | Comparative plastome study | Adds a comparative phylogenomic layer across Garcinia species relevant to chloroplast-based stress response. | [87] |
| Ripening transcriptome | RNA-seq identified active regulation of ethylene, anthocyanin, and xanthone biosynthetic genes | Provides an expression-level foundation for pathway-centered analysis; many of these genes are candidates for stress-responsive regulation. | [88] |
| eFP reference expression atlas | A reference gene-expression atlas published for mangosteen | Improves gene-expression contextualization and candidate prioritization for future stress studies. | [89] |
| SWATH-MS proteomic profiling of ripening fruit | Resolved carbohydrate-metabolism and secondary-metabolite biosynthesis networks, including ROS-scavenging enzymes and chaperones co-expressed with ripening-associated genes | First SWATH-MS quantitative proteomic resource for Garcinia fruit pericarp, offering a protein-level framework for interpreting future stress proteomics. | [90] |
| ESI-LC-MS metabolome dataset of pericarp, aril and seed | Untargeted metabolomics across ripening stages and fruit tissues catalogued xanthones, prenyl-flavonoids and osmoprotective compounds enriched in specific tissues | Provides a tissue-resolved metabolome baseline that can be re-interrogated for stress-metabolomics studies in mangosteen and related Garcinia. | [91] |
| MYB transcription factor study | In silico identification of MYB TFs linked to anthocyanin and xanthone production | Provides a candidate regulatory layer directly relevant to stress-responsive secondary metabolism. | [75] |
| Comparative tropical-fruit WRKY framework | Identified abiotic-stress- and flavonoid-associated WRKY candidates with promoter features linked to hormone and light responsiveness | Provides a comparative regulatory framework that can inform future WRKY-focused studies in Garcinia and related tropical fruit systems. | [76] |
Priority directions for Garcinia stress multi-omics emerging from the current literature include drought, heat, and salinity transcriptomes for mature leaf, root, and fruit tissues; pairing these with SWATH-style proteomics [90] and LC-MS metabolomics of xanthone- and anthocyanin-related pathways [91]; promoter-reporter and protoplast-based assays to validate candidate MYB and WRKY regulators [75,76]; and epigenomic profiling of heat and cold priming using genome and organelle resources [85,86,87] together with transcriptome and expression atlases [88,89]. Applying such approaches to G. mangostana in parallel with G. atroviridis could also yield a genus-level comparative framework linking stress acclimation to the evolution of specialized metabolism [92].
7.2 Cassava: A Tropical Staple in Transition
Cassava occupies a different but equally important position. Unlike many underutilized fruit or medicinal species, it is already a major tropical staple and has been the focus of sustained drought-related research [93,94]. Recent work on cassava increasingly combines genomics, transcriptomics, and breeding-oriented approaches to improve stress resilience, reflecting a transition from basic stress-response description toward more applied systems-level interpretation. Studies using genomic selection have shown that drought-related genomic estimated breeding values can be predicted with accuracies of 0.45–0.60 using multi-trait models incorporating transcriptome-derived priors, providing a template for omics-assisted breeding in tropical staples.
Cassava A20AN1 zinc-finger proteins have been identified as candidates that interact with stress-responsive kinases and are associated with drought and cold signaling modules, offering a testable regulatory module at the intersection of transcriptomic discovery and functional biology [43]. Combined with tissue-specific heat transcriptomics that distinguish vasculature-specific and mesophyll-specific acclimation programs [38], these studies suggest that cassava is moving toward a state where multi-layer stress data are available for more integrated interpretation, even though comprehensive proteome–transcriptome–metabolome datasets and fully validated mechanistic models remain rare [93,94].
In the context of this review, cassava therefore serves as an intermediate case between model crops and still under-resourced tropical species. It shows that tropical stress biology can become analytically stronger once genomic tools, breeding frameworks, and targeted molecular datasets begin to converge. At the same time, it illustrates a persistent challenge: even in relatively better-resourced tropical crops, the step from transcript-responsive mechanisms to robust multi-layer acclimation models is not automatic [43,93,94]. Cassava is thus best viewed as a system in transition, where the infrastructure for stronger stress multi-omics is emerging, but full integrative interpretation is still developing.
7.3 Coconut and Mango: Integrative Cold-Stress Frameworks
Coconut and mango provide stronger examples of what more clearly integrative stress-omics work can look like in tropical fruit systems. In coconut, combined transcriptomic and metabolomic analysis under cold stress has shown coordinated responses involving amino acid metabolism, lipid-associated pathways, flavonoid-related adjustment, and stress-linked regulatory pathways [62]. In mango, recent cold-stress work across cultivars with different tolerance levels similarly connected metabolite accumulation with transcriptomic changes in phytohormone signaling, MAPK-associated responses, and the ICE–CBF–COR pathway [61]. These studies are important because they move beyond simple identification of stress-responsive transcripts and instead show how biochemical and transcriptional responses can be interpreted together to explain cultivar-level differences in acclimation.
These two fruit systems are also useful because they demonstrate that tropical fruit stress biology is beginning to generate the type of integrative evidence long more common in model or temperate crops. At the same time, they highlight how limited literature still is: a few well-designed studies can now support meaningful mechanistic interpretation, but the broader field remains sparse and uneven. Coconut and mango therefore act as proof-of-concept systems showing that tropical fruit multi-omics can produce biologically rich stress-acclimation insights when studies are designed around genuine data integration rather than parallel dataset generation [61,62].
7.4 Avicennia marina and Mangrove Halophytes: Salinity Acclimation at the Tropical Coast
Mangrove systems add a different dimension because they represent tropical plants adapted to highly dynamic saline environments rather than conventional agricultural settings [65,95]. Recent work in Avicennia marina has shown that integrated transcriptome–metabolome analysis can distinguish low-salinity and high-salinity acclimation strategies, including differences in ion homeostasis, osmolyte accumulation, antioxidant metabolism, and carbon-use patterns. Low-salinity acclimation favors HAK8-mediated K+ uptake and abscisic acid (ABA) signaling, whereas high-salinity acclimation relies more heavily on Na+ sequestration, glycine betaine accumulation, and remodeling of polyphenol metabolism. Recent comparative mangrove genomics resources provide a stronger platform for these integrative analyses and enable broader comparison across true mangroves such as Kandelia obovata.
Complementary studies in mangroves have begun to resolve combined stress responses. Physiological and transcriptomic work in Kandelia obovata under cold waves has shown that antioxidant and osmotic adjustments contribute materially to stress acclimation [96]. In Kandelia obovata, combined salinity and waterlogging stress triggers distinct transcriptional reprogramming compared with either stress alone, highlighting the importance of multifactorial design in mangrove research [97]. Comparative transcriptomic and metabolomic analyses in Avicennia marina and Kandelia obovata under chilling have further identified coordinated redox- and phenylpropanoid-associated responses, underscoring mangroves’ value as comparative models for abiotic-stress biology in tropical coastal systems [98].
From a review perspective, mangroves are important not because they are underutilized crops in the agricultural sense, but because they expand the comparative range of tropical stress biology. Avicennia marina illustrates salinity-acclimation strategies in a tropical coastal halophyte [65,96], while Kandelia obovata adds combined salinity–waterlogging and chilling-stress perspectives [97,98]. These systems remind us that tropical plant acclimation cannot be adequately synthesized if attention remains restricted to a narrow group of cultivated species. They also show that ecologically specialized tropical plants can contribute biologically informative stress models, even though they are not yet fully incorporated into general stress-omics synthesis [95].
7.5 Tropical Millets: Underutilized Cereals for Climate-Resilient Multi-Omics
Millets including pearl millet (Cenchrus americanus), finger millet (Eleusine coracana), foxtail millet (Setaria italica), and proso millet (Panicum miliaceum) represent an exceptionally promising class of underutilized tropical cereals for stress multi-omics. Pearl millet already has transcriptomic, proteomic, and physiology-linked stress studies [49,52,66], while foxtail millet contributes drought- and anthocyanin-linked transcriptomic evidence [50,53]. Broader reviews of millet abiotic-stress biology reinforce the same conclusion [51]. These cereals combine intrinsic tolerance to drought, heat, and saline or marginal soils with expanding genomic resources. In pearl millet, this evidence is supported by transcriptomic and proteomic studies, while foxtail and comparative millet studies add transcriptomic and broader stress-biology context. Recent reviews and primary transcriptome studies across millet systems consistently converge on hormone signaling, reactive-oxygen-species management, osmotic adjustment, and transcription-factor reprogramming as central acclimation themes.
Pearl millet provides especially clear evidence for the value of integrating biological layers. Transcriptomic analysis under heat and drought has expanded the functional annotation landscape and identified large sets of stress-responsive genes and transcriptional regulators [49]. Proteomics and physiology then add a more mechanistic dimension: drought-focused work has highlighted superior photosynthetic maintenance, root-system adjustment, and redox buffering [52], whereas salinity-focused comparative proteomics revealed genotype-specific osmotic and ionic homeostasis together with poor correspondence between protein abundance and transcript levels [66]. In foxtail millet, drought transcriptome analyses and anthocyanin-associated transcriptomic studies have similarly linked stress acclimation to antioxidant defence, phenylpropanoid metabolism, osmotic regulation, and stress-linked regulatory networks [50,53].
Crop translational-genomics perspectives further argue that moving from candidate lists to validated tolerance alleles in orphan cereals such as pearl and finger millet will require tighter coupling of genomic selection, multi-omics candidate nomination, and high-throughput phenotyping [99]. Together, pearl millet studies support transcriptomic, proteomic, and physiology-linked stress interpretation [49,52,66], while foxtail and comparative millet work broaden this evidence base across underutilized cereals [51,53]. These studies make tropical millets a natural next step for comparative tropical stress biology beyond fruit and perennial crops.
7.6 Cross-Species Synthesis: Conserved Mechanisms, Species-Specific Responses, and Research Priorities
Despite substantial differences in phylogeny, growth form, and ecological context, a core set of molecular responses recurs consistently across the species reviewed here. ABA signaling emerges as a central regulatory hub in drought and salinity acclimation: ABA-responsive NAC and MYB transcription factors were among the most strongly induced regulators in banana under drought [36], ABA signaling modules were identified within the core DEG set shared across five stresses in oil palm [39], and HAK8-mediated ABA signaling was specifically activated under low-salinity conditions in Avicennia marina [65]. ROS buffering through superoxide dismutase, ascorbate peroxidase, and catalase was documented at the protein level in banana under salinity stress [56], confirmed through proteomic profiling in oil palm under low-temperature conditions [58], and detected at both transcript and metabolite levels in mango and coconut under cold stress [61,62]. Heat shock protein induction particularly HSP70, HSP90, and small HSPs was consistently reported under heat stress in cassava leaf and vasculature tissues [38] and within the core stress transcriptome of oil palm across multiple stress conditions [46]. Osmolyte accumulation including proline and compatible solutes was documented in mango under prolonged cold [61], in coconut seedlings under cold stress [62], and in Avicennia marina under high salinity as glycine betaine accumulation [71]. These convergent patterns suggest that the core biochemical vocabulary of abiotic stress acclimation is broadly shared across tropical lineages despite their taxonomic diversity.
Alongside these conserved mechanisms, several lineage-specific responses stand out as biologically distinctive. In Garcinia mangostana, SWATH-MS proteomic analysis revealed stress-associated remodeling of xanthone biosynthesis pathway proteins during postharvest conditions [90], while ESI-LC-MS metabolomic profiling identified prenylflavonoids and xanthone derivatives as the dominant biochemically responsive compound classes [91] a metabolic profile with no direct parallel in any other species reviewed here. In Avicennia marina, root ionomic, transcriptomic, and metabolomic integration demonstrated a switch between two distinct acclimation strategies: low salinity favored HAK8-mediated K+ uptake and ABA signaling, whereas high salinity relied on Na+ sequestration, glycine betaine accumulation, and polyphenol remodeling [65] a tiered halophytic strategy fundamentally different from the glycophytic responses of cassava or banana. In cassava, comparative leaf versus mid-vein transcriptomics identified that mesophyll tissue preferentially enhanced pyruvate synthesis under heat, while vascular tissue simultaneously remodeled sucrose–starch metabolism and phloem-associated transporters [45] a degree of within-organ metabolic partitioning not yet systematically explored in most other tropical crops. In pearl millet, integration of transcriptomics, proteomics, and physiological measurements under combined drought and heat identified root morphological plasticity and nitrogen-use coordination as key tolerance features, with 127 multi-layer-supported candidate markers identified across omics layers [57].
Several conclusions from the tropical multi-omics literature are now sufficiently supported to be stated with confidence. Transcriptomics consistently underestimates acclimation complexity: iTRAQ proteomics in banana under salinity identified ROS-scavenging, chaperone, and photosynthetic proteins as major responsive classes, with patterns that could not have been predicted from transcript data alone [56], and tissue-specific proteomic profiling in pearl millet revealed redox-buffering and photosynthetic repair proteins that were not reflected in the corresponding transcriptome [57]. Integration of transcript and metabolite layers reliably improves inference: in mango, metabolite shifts toward flavonoids, terpenoids, and amino acids helped determine the biochemical direction of cold acclimation beyond what transcriptomic ICE–CBF–COR pathway data could resolve on its own [61], while in Avicennia marina, root metabolomic data clarified which of the transcriptomic implicated pathways were biochemically enacted under each salinity regime [65]. In species with incomplete annotations, metabolomics can partially substitute for annotation gaps, as demonstrated using metabolomic profiling to validate pathway activity in oil palm despite reference incompleteness [58] and in coconut where metabolite shifts provided functional context for transcriptomic cold-response modules [62]. By contrast, most TF candidates in this literature remain associative: MYB, WRKY, NAC, and bHLH family members identified in banana [36], cacao [37], and Garcinia in silico analyses [80,81] have not been subjected to functional validation through overexpression, knockout, or protein–DNA interaction assays in the relevant tropical species. These claims should be treated as working hypotheses requiring functional testing before causal roles in acclimation can be established.
Based on multi-layer omics data availability, physiological characterization depth, and genome resource maturity, three systems emerge as best positioned for stress priming experimental design. Avicennia marina has integrated root ionomic, transcriptomic, and metabolomic data across two distinct salinity regimes [65], providing a multi-layer mechanistic baseline from which priming and epigenetic memory experiments could be designed against a well-characterized molecular reference. Pearl millet (Cenchrus americanus) has transcriptomic, proteomic, and physiological data under combined drought and heat [57], complementary physiology-integrated multi-omics from additional drought studies [70], an existing global germplasm collection with known tolerance variation, and active genomic improvement programs making it the most experimentally tractable system for molecular priming validation among the underutilized cereals reviewed here. Cassava (Manihot esculenta) has tissue-resolved transcriptomic data under heat stress [38], evidence of abiotic stress signaling through zinc-finger regulatory modules [43], and proteomic evidence of selective LEA protein and glutathione S-transferase stabilization under combined stress [59], providing a multi-layer foundation sufficient to support epigenetic profiling of candidate stress-memory loci. Extending these three systems toward explicitly priming-focused designs including repeated stress exposure, epigenomic profiling across stress cycles, and physiological performance tracking represents one of the clearest near-term research priorities in tropical stress biology.
8 Methodological Bottlenecks and Analytical Challenges
A persistent problem in tropical stress-omics is that many species still lack reference genomes of sufficient quality for dependable multi-layer interpretation. Even when assemblies are available, structural annotation often remains incomplete because plant genomes are frequently large, repetitive, heterozygous, or polyploid [21,26,29]. A 2024 analysis of crop genomes and gene prediction accuracy showed that annotation quality has direct implications for downstream proteomics, with typical peptide-to-gene mapping rates varying from 65% to over 90% depending on annotation completeness. This is especially relevant in tropical and underutilized plants, where stress studies often rely on draft genomes, incomplete gene models, or de novo transcript assemblies that are informative but not fully stable platforms for deeper multi-omics integration. Without stronger genome assemblies and better gene annotation, apparent biological gaps can easily reflect technical under-resolution rather than real absence of pathway components.
Recent pangenome work further complicates the picture in a productive way. Reviews now emphasize that single-reference genomes frequently miss structurally variable, presence–absence, and lineage-specific regions that can be important for stress-associated traits [20,26,28]. In underutilized species, where diversity is often poorly sampled and reference bias is strong, the absence of pangenomic resources means that biologically meaningful variation may remain invisible to standard transcriptomic or comparative analyses. Building a plant pangenome for a tropical species typically requires 10–30 high-quality accession genomes, which is achievable for rice and wheat but remains a stretch for orphan crops. In other words, incomplete reference frameworks do not just weaken annotation, they narrow the range of stress biology that can even be asked.
A second major bottleneck lies in stress phenotyping. Molecular datasets gain interpretive value only when they are linked to carefully measured physiological and phenotypic outcomes, yet this remains one of the weakest aspects of much plant stress literature [100,101,102]. In tropical and underutilized species, omics studies often include only limited endpoint measures, making it difficult to distinguish general stress response from meaningful acclimation [14]. Recent translational genomics perspectives stress that even with strong sequence resources, moving toward practical or mechanistic trait interpretation requires integration with robust phenotyping [103]. This is even more critical in species that are less experimentally standardized than major crops.
The challenge is not simply technical throughput. High-throughput phenotyping has advanced considerably through unmanned aerial vehicle (UAV)-mounted multispectral imaging, automated greenhouse platforms, and infrared and chlorophyll-fluorescence-based stress diagnostics, but review work in this area shows that the real difficulty lies in designing phenotyping systems that are biologically relevant, reproducible, and comparable across experiments [100,101]. Under abiotic stress, plant responses are strongly influenced by developmental stage, tissue selection, timing of sampling, and environmental context. If these variables are not aligned with molecular sampling, multi-omics datasets can become analytically rich but biologically ambiguous. In tropical species, where greenhouse control may be weaker and field realism often matters more, this problem is especially acute [45]. Standardized minimum information guidelines—analogous to MIAME or MIAPE—tailored for plant stress multi-omics would substantially improve comparability.
Metabolomics presents a distinct but equally serious challenge. A 2025 review on plant metabolomics in the multi-omics era makes clear that metabolite annotation remains one of the major bottlenecks in plant systems biology, especially when species produce lineage-specific compounds weakly represented in existing databases and spectral libraries [32]. Typical untargeted LC-MS datasets for tropical species still contain 40–70% unannotated features, and confidence levels of identification (MSI Level 1–4) are rarely reported consistently [33]. This issue is particularly relevant in tropical and underutilized plants, which often accumulate unusual or poorly catalogued specialized metabolites including xanthones (Garcinia), prenylflavonoids, lignans, and polyphenols [22]. In such cases, metabolomics can reveal clear biochemical change without always providing confident compound identity or pathway context. Emerging AI-assisted spectral-library tools and metabolite-identification platforms such as SIRIUS 5, MS2DeepScore, and MolDiscovery are beginning to close this gap, but validation in non-model tropical systems is still preliminary.
This annotation problem affects integration directly. Transcriptome–metabolome or proteome–metabolome analyses are most informative when pathway identities are reliable and metabolite assignments can be linked to specific biochemical processes [4,32,33]. In tropical systems, however, integration often occurs under conditions of incomplete metabolite confidence, incomplete genome annotation, and incomplete orthology mapping. As a result, current integrative studies are often stronger at showing coordination across layers than at demonstrating mechanism. This does not diminish their value, but it does mean that pathway-level inference in tropical non-model systems should be treated cautiously when metabolite identity, genome annotation [21,29], and cross-layer mapping remain incomplete.
Protein extraction and proteomic quantification present parallel challenges. Tropical tissues often contain high concentrations of polysaccharides, polyphenols, terpenoids, and specialized secondary metabolites that interfere with standard proteomic workflows [54,58]. TCA–acetone precipitation, phenol-based extraction, and detergent-based buffers each have trade-offs that are still not systematically benchmarked across tropical species, and data-independent acquisition (DIA) mass spectrometry, although promising, remains less widely adopted in tropical labs than data-dependent acquisition. Strengthening the proteomic pipeline for tropical species via standardized reference spectra, harmonized quantification protocols, and shared spectral libraries would substantially accelerate cross-layer interpretation.
A further challenge is that literature is still difficult to compare across species, even when omics datasets exist. Tropical and underutilized plants are often studied through isolated projects built around different stress protocols, tissues, sequencing depths, metabolomics platforms, and analytical pipelines [18,24,28]. This weakens the possibility of genuine comparative synthesis. Recent work on underutilized crop genomics argues that inclusivity in crop science depends not only on sequencing more species, but on developing more interoperable frameworks for using those data [19]. Community-level initiatives such as the African Orphan Crops Consortium, the Global Open Data for Agriculture and Nutrition network, and FAIR data practices for plant multi-omics are emerging as important structural correctives. Without stronger comparability, tropical stress multi-omics will continue to accumulate case studies faster than it builds generalizable biological understanding.
Finally, analytical infrastructure remains unevenly distributed. Computational demands of multi-omics integration including genome assembly, variant calling, multi-dimensional dimensionality reduction, and deep-learning-based metabolite annotation are substantial, and many tropical institutions operate without sustained access to high-performance computing, long-term data storage, or stable bioinformatic pipelines [30,103,104]. Cloud-based bioinformatic platforms (e.g., Galaxy, Terra, CyVerse, and the Plant Sciences Cloud) and regional consortium-based computing arrangements partially mitigate this constraint, but training and retention of bioinformaticians remain persistent challenges in tropical research ecosystems.
These methodological limitations do not imply that tropical and underutilized plant multi-omics is immature or unproductive. Rather, they define the conditions under which the field must now progress. Better genomes and stronger annotations [21,103], better-aligned phenotyping [100,101,102], more careful metabolite interpretation [32], standardized proteomic workflows, and more interoperable analytical design [24,104] are not secondary improvements; they are the main requirements for shifting the literature from fragmented molecular response profiles toward stronger frameworks of abiotic stress acclimation. Until these bottlenecks are addressed more systematically, the field will remain rich in signals but uneven in mechanism [4]. The major methodological bottlenecks constraining multi-omics interpretation in tropical and underutilized plants are summarized in Fig. 3.

Figure 3: Major methodological bottlenecks limiting stress multi-omics in tropical and underutilized plant systems. Four interacting constraints genome quality, annotation depth, phenotyping resolution, and data interoperability reduce mechanistic inference, cross-study comparability, candidate prioritization, and translational confidence.
The next phase of research on abiotic stress acclimation in tropical and underutilized plants will depend less on the continued expansion of isolated omics datasets and more on whether those datasets can be converted into robust comparative frameworks [14,16,20] and translational pipelines [103,105]. In major crops, recent work has shown that the real frontier is no longer simple data accumulation, but the coordinated use of genomic resources, high-resolution phenotyping, multimodal integration, and predictive analytics to generate biologically meaningful and agriculturally useful hypotheses. For tropical and underutilized species, this transition has only begun. Future progress will therefore depend on how effectively the field can close the gap between molecular profiling and explanatory power.
The first priority is the continued development of genome resources that are adequate for true multi-layer interpretation. In underutilized and tropical plants, fragmented or incomplete reference genomes remain a major obstacle to transcript annotation, peptide assignment, orthology inference, and comparative pathway analysis [21,26,29]. Recent work on orphan crop improvement and plant pangenomes makes clear that future gains will depend not only on sequencing representative genomes, but also on capturing structural variation, presence–absence polymorphisms, and gene-space diversity that single references cannot represent [20,28,30]. Long-read sequencing platforms (PacBio HiFi, Oxford Nanopore) and Hi-C scaffolding have reduced the cost of chromosome-level tropical crop assemblies from months to weeks, while telomere-to-telomere (T2T) assemblies are now achievable for selected tropical staples. In practice, future stress multi-omics in tropical plants should increasingly move toward pangenomic and diversity-aware frameworks rather than rely solely on a single accession or draft reference. Such a shift would make comparative stress biology more realistic and would reduce the risk of missing lineage-specific stress-associated genes and regulatory elements.
The second priority is to improve spatial and cellular resolution in stress studies. One of the major limitations of current tropical stress multi-omics is that most datasets are still generated from bulk tissues, which inevitably compress cellular heterogeneity into averaged profiles [106,107]. Recent reviews on single-cell and spatial omics in plants show that these technologies can resolve cell-type-specific dynamics [108], tissue-level organization [109], and the spatial logic of stress responses [110]. A 2024 best-practices review on single-cell RNA sequencing (scRNA-seq) emphasized that protoplast isolation efficiency, transcript dropout rates, and cell-type annotation consistency are key methodological parameters that must be reported transparently for reproducibility. Applications in rice, maize, and Arabidopsis have already revealed cell-type-specific stress-response atlases that reconfigure our understanding of drought, salinity, and heat acclimation. In the context of abiotic stress acclimation, this matters because many key processes including ion transport, vascular signaling, meristem maintenance, and localized antioxidant responses are unlikely to be understood fully from bulk RNA or metabolite profiles alone.
Spatial transcriptomics and emerging spatial metabolomics approaches for example, matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI) and desorption electrospray ionization mass spectrometry imaging (DESI-MSI) further extend this logic by mapping expression and metabolite distributions in situ, enabling interpretation of stress responses in their anatomical context [109,110]. For tropical and underutilized plants, the immediate goal may not be routine single-cell deployment across all species, but rather the gradual introduction of these approaches in strategically chosen systems such as cassava roots under drought [106], banana leaves under combined heat and drought [107], or A. marina root tissues under fluctuating salinity, where they can reveal stress-response architecture masked by bulk omics.
A third direction is methodological rather than purely experimental. As omics datasets increase in size and complexity, integrative analysis will increasingly depend on machine-learning frameworks capable of extracting cross-layer patterns from genomic, transcriptomic, metabolomic, and phenotypic data [30,105,111]. Recent work suggests that these tools may be especially useful in resource-limited species because transfer learning and multimodal ranking can help prioritize candidates where direct validation capacity remains limited. Transfer learning from Arabidopsis or rice stress datasets may therefore be useful, but only if models are recalibrated carefully for tropical species with distinct genomes, metabolites, and ecological contexts.
This direction is especially relevant in tropical and underutilized plants, where sparse datasets, incomplete annotations, and small sample sizes often limit conventional integration. In this setting, AI-assisted tools are best used for feature ranking, transfer of information from better-annotated crops, and cross-layer pattern discovery, but only when predictions remain interpretable and are tested against independent biological evidence [30,105,111]. The future of tropical stress multi-omics therefore lies not simply in adopting AI as a fashionable add-on, but in using it to strengthen candidate prioritization, comparative interpretation, and biologically grounded hypothesis generation.
The fourth priority is to develop validation-oriented experimental pipelines. Many tropical stress omics datasets currently end at candidate nomination, with limited follow-up functional work [4,10,35]. Stress-focused validation should combine transient expression systems and stable transformation where feasible [14], CRISPR/Cas9-based editing and emerging base- or prime-editing tools [112], and natural-variation panels such as cassava GWAS populations [94] and pearl millet association panels [99]. These approaches would move tropical stress studies from candidate prioritization toward functional confirmation, especially where transformation remains inefficient.
The fifth priority is conceptual. Much of the existing literature still treats omics as a way to identify stress signatures, whereas the next phase should use multi-omics to define translationally relevant mechanisms. This means linking molecular responses to measurable physiological traits, candidate regulators, and eventually to selection or improvement strategies [14,103,105]. In underutilized crops especially, recent reviews argue that genomics and associated computational methods should no longer be viewed only as descriptive tools but as part of broader pathways toward crop improvement, diversification, and resilience [19,28,30]. For tropical systems, this will require a stronger culture of experimental comparability, more meaningful phenotyping, and a clearer pipeline from candidate identification to functional testing. Without that transition, the field risks remaining rich in omics data but poor in mechanistic and translational synthesis.
The final priority is intellectual rather than technical. Tropical and underutilized plants should not be treated simply as extensions of model-crop biology, but as systems capable of contributing their own comparative insights into stress acclimation. As work on underutilized crops increasingly shows, these species are important not only for resilience and diversification in agri-food systems, but also because they expand the biological range through which adaptation, metabolic plasticity, and stress-response diversity can be understood [19,24,28]. Future multi-omics research will be most valuable when it does not merely transfer tools from major crops to minor species, but uses these species to broaden the conceptual foundation of plant stress biology itself. In this sense, the future of tropical and underutilized plant multi-omics is not peripheral to the field; it is central to making that field more biologically inclusive and more relevant to global agricultural realities [30]. These interconnected five priorities are illustrated in Fig. 4.

Figure 4: Future directions for multi-omics in tropical and underutilized plant stress biology. Pangenomic genome resources are linked to broader crop-diversity and pangenome framework. Single-cell and spatial omics, AI-assisted multimodal integration, validation-oriented experimental pipelines, and translational payoff are shown as mutually reinforcing directions rather than isolated tasks.
Across the tropical and underutilized species examined here, convergent multi-omics evidence points to ABA signaling, ROS buffering, heat shock protein induction, and osmolyte accumulation as conserved acclimation mechanisms, while lineage-specific responses in Garcinia mangostana, Avicennia marina, cassava, and pearl millet highlight the additional biochemical diversity that a broader comparative framework can reveal.
At the same time, the review also shows that the field remains constrained by incomplete reference genomes, inconsistent phenotyping, limited metabolite annotation, uneven validation pipelines, and weak cross-study comparability. These limitations do not diminish the value of existing work, but they do explain why many studies remain stronger at identifying stress-associated signatures than at resolving regulatory mechanisms. Future progress will require stronger genome and pangenome resources, better-aligned experimental design, and more rigorous cross-layer integration. It will also require greater emphasis on regulatory and epigenetic interpretation and deliberate adoption of single-cell, spatial, and AI-assisted framework.
The broader significance of this field is not only agricultural but conceptual. Tropical and underutilized plants should not be treated merely as peripheral additions to model-crop biology. They represent biologically informative systems capable of expanding current understanding of stress acclimation, metabolic plasticity, and adaptive diversity. A more inclusive multi-omics framework one that places these species more centrally within comparative stress research and pangenome-aware crop biology will improve not only the study of resilient crops, but also the broader biological interpretation of plant acclimation under environmental change.
Acknowledgement:
Funding Statement: HHG is supported by UKM Research University Grant (GUP-2024-092), YPASM Research Grant, and Malaysian Ministry of Higher Education Fundamental Research Grant (FRGS/1/2024/STG01/UKM/02/5).
Author Contributions: The authors confirm contribution to the paper as follows: study conception and review framing: Saqib Ali, Hoe-Han Goh; literature evaluation and draft manuscript preparation: Saqib Ali; critical revision, supervision, strategic input, and corresponding authorship: Hoe-Han Goh. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: This is a review article. All data discussed are available through the cited literature. No new primary datasets were generated for this review.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest.
References
1. Zhang H , Zhu J , Gong Z , Zhu JK . Abiotic stress responses in plants. Nat Rev Genet. 2022; 23( 2): 104– 19. doi:10.1038/s41576-021-00413-0. [Google Scholar] [CrossRef]
2. Jiang Z , van Zanten M , Sasidharan R . Mechanisms of plant acclimation to multiple abiotic stresses. Commun Biol. 2025; 8: 655. doi:10.1038/s42003-025-08077-w. [Google Scholar] [CrossRef]
3. Waadt R , Seller CA , Hsu PK , Takahashi Y , Munemasa S , Schroeder JI . Plant hormone regulation of abiotic stress responses. Nat Rev Mol Cell Biol. 2022; 23( 10): 680– 94. doi:10.1038/s41580-022-00479-6. [Google Scholar] [CrossRef]
4. Jamil IN , Remali J , Azizan KA , Nor Muhammad NA , Arita M , Goh HH , et al. Systematic multi-omics integration (MOI) approach in plant systems biology. Front Plant Sci. 2020; 11: 944. doi:10.3389/fpls.2020.00944. [Google Scholar] [CrossRef]
5. Pascual LS , Serna E , Ghani A , Lyu Z , Immadi MS , Joshi T , et al. Multi-omics-based insights into tomato adaptation to multifactorial stress combination. Plant Physiol. 2025; 199( 3): kiaf519. doi:10.1093/plphys/kiaf519. [Google Scholar] [CrossRef]
6. Ngara R , Shavrukov Y . Omics and multi-omics insights into plant responses to abiotic stresses. Plants. 2026; 15( 5): 799. doi:10.3390/plants15050799. [Google Scholar] [CrossRef]
7. Saleem MH , Noreen S , Ishaq I , Saleem A , Ali Khan K , Ercisli S , et al. Omics technologies: unraveling abiotic stress tolerance mechanisms for sustainable crop improvement. J Plant Growth Regul. 2025; 44( 7): 4165– 87. doi:10.1007/s00344-025-11674-y. [Google Scholar] [CrossRef]
8. Munir N , Hanif M , Abideen Z , Sohail M , El-Keblawy A , Radicetti E , et al. Mechanisms and strategies of plant microbiome interactions to mitigate abiotic stresses. Agronomy. 2022; 12( 9): 2069. doi:10.3390/agronomy12092069. [Google Scholar] [CrossRef]
9. Varshney RK , Nayak SN , May GD , Jackson SA . Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009; 27( 9): 522– 30. doi:10.1016/j.tibtech.2009.05.006. [Google Scholar] [CrossRef]
10. Choudhary A , Verma H , Pandey A , Ali A , Singh SS . Multi-omics approaches to decipher abiotic stress tolerance in plants. J Plant Physiol. 2025; 299: 154430. doi:10.1016/j.jplph.2025.154430. [Google Scholar] [CrossRef]
11. Varadharajan V , Rajendran R , Muthuramalingam P , Runthala A , Madhesh V , Swaminathan G , et al. Multi-omics approaches against abiotic and biotic stress—a review. Plants. 2025; 14( 6): 865. doi:10.3390/plants14060865. [Google Scholar] [CrossRef]
12. Aizat WM , Ismail I , Noor NM . Recent development in omics studies. In: Omics applications for systems biology. Cham, Switzerland: Springer; 2018. p. 1– 9. doi:10.1007/978-3-319-98758-3_1. [Google Scholar] [CrossRef]
13. Zagorščak M , Abdelhakim L , Rodriguez-Granados NY , Blejec A , Ramšak Ž , Zrimec J , et al. Integrative multi-omics analysis reveals distinct and stress-shared molecular responses in potato under heat, drought, and combined heat–drought–waterlogging. Plant Physiol. 2025; 197( 4): kiaf126. doi:10.1093/plphys/kiaf126. [Google Scholar] [CrossRef]
14. Dakal TC , Dagariya S , Goswami B , Rathore R , Rankawat R , Bhargavi HA , et al. Integrative multi-omics approaches for crop abiotic stress tolerance. Discover Plants. 2025; 2( 1): 361. doi:10.1007/s44372-025-00431-w. [Google Scholar] [CrossRef]
15. Raza A , Tabassum J , Fakhar AZ , Sharif R , Chen H , Zhang C , et al. Smart reprograming of plants against salinity stress using modern biotechnological tools. Crit Rev Biotechnol. 2023; 43( 7): 1035– 62. doi:10.1080/07388551.2022.2093695. [Google Scholar] [CrossRef]
16. Yang Y , Ali Saand M , Huang L , Abdelaal WB , Zhang J , Wu Y , et al. Applications of multi-omics technologies for crop improvement. Front Plant Sci. 2021; 12: 563953. doi:10.3389/fpls.2021.563953. [Google Scholar] [CrossRef]
17. Ding T , Li W , Li F , Ren M , Wang W . microRNAs: key regulators in plant responses to abiotic and biotic stresses via endogenous and cross-Kingdom mechanisms. Int J Mol Sci. 2024; 25( 2): 1154. doi:10.3390/ijms25021154. [Google Scholar] [CrossRef]
18. Ndlovu M , Scheelbeek P , Ngidi M , Mabhaudhi T . Underutilized crops for diverse, resilient and healthy agri-food systems: a systematic review of sub-Saharan Africa. Front Sustain Food Syst. 2024; 8: 1498402. doi:10.3389/fsufs.2024.1498402. [Google Scholar] [CrossRef]
19. Akpojotor U , Oluwole O , Oyatomi O , Paliwal R , Abberton M . Research and developmental strategies to hasten the improvement of orphan crops. GM Crops Food. 2025; 16( 1): 46– 71. doi:10.1080/21645698.2024.2423987. [Google Scholar] [CrossRef]
20. Schreiber M , Jayakodi M , Stein N , Mascher M . Plant pangenomes for crop improvement, biodiversity and evolution. Nat Rev Genet. 2024; 25( 8): 563– 77. doi:10.1038/s41576-024-00691-4. [Google Scholar] [CrossRef]
21. Bernal-Gallardo JJ , de Folter S . Plant genome information facilitates plant functional genomics. Planta. 2024; 259( 5): 117. doi:10.1007/s00425-024-04397-z. [Google Scholar] [CrossRef]
22. Ali A , Bhattacharjee B . Nutrition security, constraints, and agro-diversification strategies of neglected and underutilized crops to fight global hidden hunger. Front Nutr. 2023; 10: 1144439. doi:10.3389/fnut.2023.1144439. [Google Scholar] [CrossRef]
23. Roychowdhury R , Das SP , Gupta A , Parihar P , Chandrasekhar K , Sarker U , et al. Multi-omics pipeline and omics-integration approach to decipher plant’s abiotic stress tolerance responses. Genes. 2023; 14( 6): 1281. doi:10.3390/genes14061281. [Google Scholar] [CrossRef]
24. Shorinola O , Marks R , Emmrich P , Jones C , Odeny D , Chapman MA . Integrative and inclusive genomics to promote the use of underutilised crops. Nat Commun. 2024; 15( 1): 320. doi:10.1038/s41467-023-44535-x. [Google Scholar] [CrossRef]
25. Shao J , Huang K , Batool M , Idrees F , Afzal R , Haroon M , et al. Versatile roles of polyamines in improving abiotic stress tolerance of plants. Front Plant Sci. 2022; 13: 1003155. doi:10.3389/fpls.2022.1003155. [Google Scholar] [CrossRef]
26. MacNish TR , Danilevicz MF , Bayer PE , Bestry MS , Edwards D . Application of machine learning and genomics for orphan crop improvement. Nat Commun. 2025; 16: 982. doi:10.1038/s41467-025-56330-x. [Google Scholar] [CrossRef]
27. Considine MJ , Foyer CH . Oxygen and reactive oxygen species-dependent regulation of plant growth and development. Plant Physiol. 2021; 186( 1): 79– 92. doi:10.1093/plphys/kiaa077. [Google Scholar] [CrossRef]
28. Hu H , Zhao J , Thomas WJW , Batley J , Edwards D . The role of pangenomics in orphan crop improvement. Nat Commun. 2025; 16: 118. doi:10.1038/s41467-024-55260-4. [Google Scholar] [CrossRef]
29. Abbas Q , Wilhelm M , Kuster B , Poppenberger B , Frishman D . Exploring crop genomes: assembly features, gene prediction accuracy, and implications for proteomics studies. BMC Genom. 2024; 25( 1): 619. doi:10.1186/s12864-024-10521-w. [Google Scholar] [CrossRef]
30. Vasupalli N , Bhat JA , Jain P , Sri T , Islam MA , Shivaraj SM , et al. Omics big data for crop improvement: opportunities and challenges. Crop J. 2024; 12( 6): 1517– 32. doi:10.1016/j.cj.2024.10.007. [Google Scholar] [CrossRef]
31. Talabi AO , Vikram P , Thushar S , Rahman H , Ahmadzai H , Nhamo N , et al. Orphan crops: a best fit for dietary enrichment and diversification in highly deteriorated marginal environments. Front Plant Sci. 2022; 13: 839704. doi:10.3389/fpls.2022.839704. [Google Scholar] [CrossRef]
32. Hao Y , Zhang Z , Luo E , Yang J , Wang S . Plant metabolomics: applications and challenges in the era of multi-omics big data. aBIOTECH. 2025; 6( 1): 116– 32. doi:10.1007/s42994-024-00194-0. [Google Scholar] [CrossRef]
33. Xu Y , Fu X . Reprogramming of plant central metabolism in response to abiotic stresses: a metabolomics view. Int J Mol Sci. 2022; 23( 10): 5716. doi:10.3390/ijms23105716. [Google Scholar] [CrossRef]
34. Galili G , Amir R , Fernie AR . The regulation of essential amino acid synthesis and accumulation in plants. Annu Rev Plant Biol. 2016; 67: 153– 78. doi:10.1146/annurev-arplant-043015-112213. [Google Scholar] [CrossRef]
35. Lamsaadi N , Maarouf O , Lahmaoui S , Msaad H , Farssi O , Hamim C , et al. Key plant transcription factors in crop tolerance to abiotic stresses. Phyton-Int J Exp Bot. 2025; 94( 11): 3585– 610. doi:10.32604/phyton.2025.072311. [Google Scholar] [CrossRef]
36. Muthusamy M , Uma S , Backiyarani S , Saraswathi MS , Chandrasekar A . Transcriptomic changes of drought-tolerant and sensitive banana cultivars exposed to drought stress. Front Plant Sci. 2016; 7: 1609. doi:10.3389/fpls.2016.01609. [Google Scholar] [CrossRef]
37. Osorio Zambrano MA , Rodríguez Pérez L , Papatheodorou I , Terán W . All roads lead to Rome: integrated physiological and transcriptomic analysis of cacao drought response reveals different ways to achieve tolerance in two hybrid clones. Front Plant Sci. 2026; 17: 1764400. doi:10.3389/fpls.2026.1764400. [Google Scholar] [CrossRef]
38. Wang S , Zhou X , Pan K , Zhang H , Shen X , Luo J , et al. Distinct heat response molecular mechanisms emerge in cassava vasculature compared to leaf mesophyll tissue under high temperature stress. Front Plant Sci. 2023; 14: 1281436. doi:10.3389/fpls.2023.1281436. [Google Scholar] [CrossRef]
39. Lee FC , Yeap WC , Kee SY , Kulaveerasingam H , Ross Appleton D . Core transcriptome network modulates temperature (heat and cold) and osmotic (drought, salinity, and waterlogging) stress responses in oil palm. Front Plant Sci. 2024; 15: 1497017. doi:10.3389/fpls.2024.1497017. [Google Scholar] [CrossRef]
40. Wei Y , Shi J , Xie X , Zhang F , Dong H , Li Y , et al. Transcriptome sequence reveal the roles of MaGME777 and MabHLH770 in drought tolerance in Musa acuminata. Plant Sci. 2025; 356: 112495. doi:10.1016/j.plantsci.2025.112495. [Google Scholar] [CrossRef]
41. Xu Y , Hu W , Liu J , Zhang J , Jia C , Miao H , et al. A banana aquaporin gene, MaPIP1;1, is involved in tolerance to drought and salt stresses. BMC Plant Biol. 2014; 14( 1): 59. doi:10.1186/1471-2229-14-59. [Google Scholar] [CrossRef]
42. Yu X , Liu Z , Sun X . Single-cell and spatial multi-omics in the plant sciences: technical advances, applications, and perspectives. Plant Commun. 2023; 4( 3): 100508. doi:10.1016/j.xplc.2022.100508. [Google Scholar] [CrossRef]
43. Su Y , Chen YL , Wu YL , Fan XW , Li YZ . Three cassava A20/AN1 family genes, Metip3 (5, and 7), can bestow on tolerance of plants to multiple abiotic stresses but show functional convergence and divergence. Plant Sci. 2024; 346: 112163. doi:10.1016/j.plantsci.2024.112163. [Google Scholar] [CrossRef]
44. Yow AG , Laosuntisuk K , Young RA , Doherty CJ , Gillitt N , Perkins-Veazie P , et al. Comparative transcriptome analysis reveals candidate genes for cold stress response and early flowering in pineapple. Sci Rep. 2023; 13( 1): 18890. doi:10.1038/s41598-023-45722-y. [Google Scholar] [CrossRef]
45. Lim H , Kobayashi MJ , Marsoem SN , Irawati D , Kosugi A , Kondo T , et al. Transcriptomic responses of oil palm (Elaeis guineensis) stem to waterlogging at plantation in relation to precipitation seasonality. Front Plant Sci. 2023; 14: 1213496. doi:10.3389/fpls.2023.1213496. [Google Scholar] [CrossRef]
46. Sasidharan R , Schippers JHM , Schmidt RR . Redox and low-oxygen stress: signal integration and interplay. Plant Physiol. 2021; 186( 1): 66– 78. doi:10.1093/plphys/kiaa081. [Google Scholar] [CrossRef]
47. Gamboa-Tuz SD , Pereira-Santana A , Zamora-Briseño JA , Castano E , Espadas-Gil F , Ayala-Sumuano JT , et al. Transcriptomics and co-expression networks reveal tissue-specific responses and regulatory hubs under mild and severe drought in Papaya (Carica papaya L.). Sci Rep. 2018; 8( 1): 14539. doi:10.1038/s41598-018-32904-2. [Google Scholar] [CrossRef]
48. Estrella-Maldonado H , Ramírez AG , Ortiz GF , Peraza-Echeverría S , Martínez-de la Vega O , Góngora-Castillo E , et al. Transcriptomic analysis reveals key transcription factors associated to drought tolerance in a wild Papaya (Carica papaya) genotype. PLoS One. 2021; 16( 1): e0245855. doi:10.1371/journal.pone.0245855. [Google Scholar] [CrossRef]
49. Saini DK , Chopra Y , Singh J , Sandhu KS , Kumar A , Bazzer S , et al. Comprehensive evaluation of mapping complex traits in wheat using genome-wide association studies. Mol Breeding. 2022; 42: 1. doi:10.1007/s11032-021-01272-7. [Google Scholar] [CrossRef]
50. Wang J , Sun Z , Wang X , Tang Y , Li X , Ren C , et al. Transcriptome-based analysis of key pathways relating to yield formation stage of foxtail millet under different drought stress conditions. Front Plant Sci. 2022; 13: 1110910. doi:10.3389/fpls.2022.1110910. [Google Scholar] [CrossRef]
51. Saleem S , Ul Mushtaq N , Hafiz Shah W , Rasool A , Rehman Hakeem K , Ul Rehman R . Morpho-physiological, biochemical and molecular adaptation of millets to abiotic stresses: a review. Phyton. 2021; 90( 5): 1363– 85. doi:10.32604/phyton.2021.014826. [Google Scholar] [CrossRef]
52. Ghatak A , Chaturvedi P , Bachmann G , Valledor L , Ramšak Ž , Bazargani MM , et al. Physiological and proteomic signatures reveal mechanisms of superior drought resilience in pearl millet compared to wheat. Front Plant Sci. 2020; 11: 600278. doi:10.3389/fpls.2020.600278. [Google Scholar] [CrossRef]
53. Zhao Y , Li Y , Zhen X , Zhang J , Zhang Q , Liu Z , et al. Uncovering the mechanism of anthocyanin accumulation in a purple-leaved variety of foxtail millet (Setaria italica) by transcriptome analysis. PeerJ. 2022; 10: e14099. doi:10.7717/peerj.14099. [Google Scholar] [CrossRef]
54. Kosová K , Vítámvás P , Urban MO , Prášil IT , Renaut J . Plant abiotic stress proteomics: the major factors determining alterations in cellular proteome. Front Plant Sci. 2018; 9: 122. doi:10.3389/fpls.2018.00122. [Google Scholar] [CrossRef]
55. Salam U , Ullah S , Tang ZH , Elateeq AA , Khan Y , Khan J , et al. Plant metabolomics: an overview of the role of primary and secondary metabolites against different environmental stress factors. Life. 2023; 13( 3): 706. doi:10.3390/life13030706. [Google Scholar] [CrossRef]
56. Ji FS , Tang L , Li YY , Wang WC , Yang Z , Li XG , et al. Differential proteomic analysis reveals the mechanism of Musa paradisiaca responding to salt stress. Mol Biol Rep. 2019; 46( 1): 1057– 68. doi:10.1007/s11033-018-4564-2. [Google Scholar] [CrossRef]
57. Ghatak A , Chaturvedi P , Nagler M , Roustan V , Lyon D , Bachmann G , et al. Comprehensive tissue-specific proteome analysis of drought stress responses in Pennisetum glaucum (L.) R. Br. (Pearl millet). J Proteom. 2016; 143: 122– 35. doi:10.1016/j.jprot.2016.02.032. [Google Scholar] [CrossRef]
58. John Martin JJ , Song Y , Hou M , Zhou L , Liu X , Li X , et al. Multi-omics approaches in oil palm research: a comprehensive review of metabolomics, proteomics, and transcriptomics based on low-temperature stress. Int J Mol Sci. 2024; 25( 14): 7695. doi:10.3390/ijms25147695. [Google Scholar] [CrossRef]
59. Zhao P , Liu P , Shao J , Li C , Wang B , Guo X , et al. Analysis of different strategies adapted by two cassava cultivars in response to drought stress: ensuring survival or continuing growth. J Exp Bot. 2015; 66( 5): 1477– 88. doi:10.1093/jxb/eru507. [Google Scholar] [CrossRef]
60. Chevilly S , Dolz-Edo L , Martínez-Sánchez G , Morcillo L , Vilagrosa A , López-Nicolás JM , et al. Distinctive traits for drought and salt stress tolerance in melon (Cucumis melo L.). Front Plant Sci. 2021; 12: 777060. doi:10.3389/fpls.2021.777060. [Google Scholar] [CrossRef]
61. Kong Y , Hou X , Liu Z , Li Y . Cold-stress induced metabolomic and transcriptomic changes in leaves of three mango varieties with different cold tolerance. BMC Plant Biol. 2024; 24( 1): 266. doi:10.1186/s12870-024-04983-z. [Google Scholar] [CrossRef]
62. Lu L , Yang W , Dong Z , Tang L , Liu Y , Xie S , et al. Integrated transcriptomic and metabolomics analyses reveal molecular responses to cold stress in coconut (Cocos nucifera L.) seedlings. Int J Mol Sci. 2023; 24( 19): 14563. doi:10.3390/ijms241914563. [Google Scholar] [CrossRef]
63. Subramanian I , Verma S , Kumar S , Jere A , Anamika K . Multi-omics data integration, interpretation, and its application. Bioinform Biol Insights. 2020; 14: 1– 24. doi:10.1177/1177932219899051. [Google Scholar] [CrossRef]
64. Argelaguet R , Arnol D , Bredikhin D , Deloro Y , Velten B , Marioni JC , et al. MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol. 2020; 21( 1): 111. doi:10.1186/s13059-020-02015-1. [Google Scholar] [CrossRef]
65. Li J , Xu CQ , Song LY , Guo ZJ , Zhang LD , Tang HC , et al. Integrative analysis of transcriptome and metabolome reveal the differential tolerance mechanisms to low and high salinity in the roots of facultative halophyte Avicennia marina. Tree Physiol. 2024; 44( 8): tpae082. doi:10.1093/treephys/tpae082. [Google Scholar] [CrossRef]
66. Jha S , Maity S , Singh J , Chouhan C , Tak N , Ambatipudi K . Integrated physiological and comparative proteomics analysis of contrasting genotypes of pearl millet reveals underlying salt-responsive mechanisms. Physiol Plant. 2022; 174: e13605. doi:10.1111/ppl.13605. [Google Scholar] [CrossRef]
67. Raza A , Li Y , Prakash CS , Hu Z . Panomics to manage combined abiotic stresses in plants. Trends Plant Sci. 2025; 30( 10): 1079– 84. doi:10.1016/j.tplants.2025.03.001. [Google Scholar] [CrossRef]
68. Mejía-Alvarado FS , Caicedo-Zambrano AF , Botero-Rozo D , Araque L , Bayona-Rodríguez CJ , Jazayeri SM , et al. Integrative analysis of transcriptomic profiles and physiological responses provide new insights into drought stress tolerance in oil palm (Elaeis guineensis Jacq.). Int J Mol Sci. 2024; 25( 16): 8761. doi:10.3390/ijms25168761. [Google Scholar] [CrossRef]
69. Xu WB , Cao F , Liu P , Yan K , Guo QH . The multifaceted role of RNA-based regulation in plant stress memory. Front Plant Sci. 2024; 15: 1387575. doi:10.3389/fpls.2024.1387575. [Google Scholar] [CrossRef]
70. Yoon Y , Seo DH , Shin H , Kim HJ , Kim CM , Jang G . The role of stress-responsive transcription factors in modulating abiotic stress tolerance in plants. Agronomy. 2020; 10( 6): 788. doi:10.3390/agronomy10060788. [Google Scholar] [CrossRef]
71. Gaude AA , Siqueira RH , Botelho SB , Jalmi SK . Epigenetic arsenal for stress mitigation in plants. Biochim Biophys Acta BBA Gen Subj. 2024; 1868( 7): 130620. doi:10.1016/j.bbagen.2024.130620. [Google Scholar] [CrossRef]
72. Ueda M , Seki M . Histone modifications form epigenetic regulatory networks to regulate abiotic stress response. Plant Physiol. 2020; 182( 1): 15– 26. doi:10.1104/pp.19.00988. [Google Scholar] [CrossRef]
73. Manna M , Thakur T , Chirom O , Mandlik R , Deshmukh R , Salvi P . Transcription factors as key molecular target to strengthen the drought stress tolerance in plants. Physiol Plant. 2021; 172( 2): 847– 68. doi:10.1111/ppl.13268. [Google Scholar] [CrossRef]
74. Palapol Y , Ketsa S , Kui LW , Ferguson IB , Allan AC . A MYB transcription factor regulates anthocyanin biosynthesis in mangosteen (Garcinia mangostana L.) fruit during ripening. Planta. 2009; 229( 6): 1323– 34. doi:10.1007/s00425-009-0917-3. [Google Scholar] [CrossRef]
75. Wee CC , Arita M , Ramlee IA , Muhammad NAN , Subbiah VK , Goh HH . In silico identification of MYB transcription factors involved in mangosteen anthocyanin and xanthone production. Trop Plant Biol. 2026; 19( 1): 1. doi:10.1007/s12042-025-09457-6. [Google Scholar] [CrossRef]
76. Ma F , Zhou H , Yang H , Huang D , Xing W , Wu B , et al. WRKY transcription factors in passion fruit analysis reveals key PeWRKYs involved in abiotic stress and flavonoid biosynthesis. Int J Biol Macromol. 2024; 256: 128063. doi:10.1016/j.ijbiomac.2023.128063. [Google Scholar] [CrossRef]
77. Roszelin S‘M , Japlus KN , Goh HH , Md Isa N . Regulation of OsSAP8 promoter in response to abiotic stresses. Malays Appl Biol. 2025; 54( 3): 46– 58. doi:10.55230/mabjournal.v54i3.3210. [Google Scholar] [CrossRef]
78. Junaid MD , Chaudhry UK , Şanlı BA , Gökçe AF , Öztürk ZN . A review of the potential involvement of small RNAs in transgenerational abiotic stress memory in plants. Funct Integr Genom. 2024; 24( 2): 74. doi:10.1007/s10142-024-01354-7. [Google Scholar] [CrossRef]
79. Siddique AB , Parveen S , Rahman MZ , Rahman J . Revisiting plant stress memory: mechanisms and contribution to stress adaptation. Physiol Mol Biol Plants. 2024; 30( 2): 349– 67. doi:10.1007/s12298-024-01422-z. [Google Scholar] [CrossRef]
80. Ma X , Zhao F , Zhou B . The characters of non-coding RNAs and their biological roles in plant development and abiotic stress response. Int J Mol Sci. 2022; 23( 8): 4124. doi:10.3390/ijms23084124. [Google Scholar] [CrossRef]
81. Yang H , Cui Y , Feng Y , Hu Y , Liu L , Duan L . Long non-coding RNAs of plants in response to abiotic stresses and their regulating roles in promoting environmental adaption. Cells. 2023; 12( 5): 729. doi:10.3390/cells12050729. [Google Scholar] [CrossRef]
82. Sharma M , Sidhu AK , Samota MK , Gupta M , Koli P , Choudhary M . Post-translational modifications in histones and their role in abiotic stress tolerance in plants. Proteomes. 2023; 11( 4): 38. doi:10.3390/proteomes11040038. [Google Scholar] [CrossRef]
83. Zheng S , Zhao W , Liu Z , Geng Z , Li Q , Liu B , et al. Establishment and maintenance of heat-stress memory in plants. Int J Mol Sci. 2024; 25( 16): 8976. doi:10.3390/ijms25168976. [Google Scholar] [CrossRef]
84. He Y , Li Z . Epigenetic environmental memories in plants: establishment, maintenance, and reprogramming. Trends Genet. 2018; 34( 11): 856– 66. doi:10.1016/j.tig.2018.07.006. [Google Scholar] [CrossRef]
85. Midin MR , Goh HH . The mangosteen genome. In: Chapman MA , editor. Underutilised crop genomes. Cham, Switzerland: Springer; 2022. p. 111– 29. doi:10.1007/978-3-031-00848-1_7. [Google Scholar] [CrossRef]
86. Wee CC , Nor Muhammad NA , Subbiah VK , Arita M , Nakamura Y , Goh HH . Mitochondrial genome of Garcinia mangostana L. variety Mesta. Sci Rep. 2022; 12( 1): 9480. doi:10.1038/s41598-022-13706-z. [Google Scholar] [CrossRef]
87. Wee CC , Nor Muhammad NA , Subbiah VK , Arita M , Nakamura Y , Goh HH . Plastomes of Garcinia mangostana L. and comparative analysis with other Garcinia species. Plants. 2023; 12( 4): 930. doi:10.3390/plants12040930. [Google Scholar] [CrossRef]
88. Jamil IN , Abdul-Rahman A , Goh HH , Aizat WM . Transcriptomics analysis of mangosteen ripening revealed active regulation of ethylene, anthocyanin and xanthone biosynthetic genes. Postharvest Biol Technol. 2023; 198: 112257. doi:10.1016/j.postharvbio.2023.112257. [Google Scholar] [CrossRef]
89. Wee CC , Pasha A , Provart NJ , Nor Muhammad NA , Subbiah VK , Arita M , et al. An eFP reference gene expression atlas for mangosteen. Sci Hortic. 2024; 327: 112846. doi:10.1016/j.scienta.2024.112846. [Google Scholar] [CrossRef]
90. Jamil IN , Sanusi S , Mackeen MM , Noor NM , Aizat WM . SWATH-MS proteomics and postharvest analyses of mangosteen ripening revealed intricate regulation of carbohydrate metabolism and secondary metabolite biosynthesis. Postharvest Biol Technol. 2021; 176: 111493. doi:10.1016/j.postharvbio.2021.111493. [Google Scholar] [CrossRef]
91. Mamat SF , Azizan KA , Baharum SN , Noor NM , Aizat WM . ESI-LC-MS based-metabolomics data of mangosteen (Garcinia mangostana Linn.) fruit pericarp, aril and seed at different ripening stages. Data Brief. 2018; 17: 1074– 7. doi:10.1016/j.dib.2018.02.033. [Google Scholar] [CrossRef]
92. Shahid M , Law D , Azfaralariff A , Mackeen MM , Chong TF , Fazry S . Phytochemicals and biological activities of Garcinia atroviridis: a critical review. Toxics. 2022; 10( 11): 656. doi:10.3390/toxics10110656. [Google Scholar] [CrossRef]
93. da Costa WG , Bandeira E Souza M , Azevedo CF , Nascimento M , Morgante CV , Borel JC , et al. Optimizing drought tolerance in cassava through genomic selection. Front Plant Sci. 2024; 15: 1483340. doi:10.3389/fpls.2024.1483340. [Google Scholar] [CrossRef]
94. Charles O . A review of drought-stress responsive genes and their applications for drought stress tolerance in cassava (Manihot esculenta Crantz). Discover Biotechnol. 2024; 1( 1): 5. doi:10.1007/s44340-024-00006-7. [Google Scholar] [CrossRef]
95. Yuan J , Gu W , Yang H , Wang M . Comparative and phylogenetic analyses of the chloroplast genomes of mangrove plants. Sci Rep. 2025; 15( 1): 3915. doi:10.1038/s41598-024-81894-x. [Google Scholar] [CrossRef]
96. He S , Wang X , Du Z , Liang P , Zhong Y , Wang L , et al. Physiological and transcriptomic responses to cold waves of the most cold-tolerant mangrove, Kandelia obovata. Front Plant Sci. 2023; 14: 1069055. doi:10.3389/fpls.2023.1069055. [Google Scholar] [CrossRef]
97. Liu H , An X , Liu X , Yang S , Liu Y , Wei X , et al. Molecular mechanism of salinity and waterlogging tolerance in mangrove Kandelia obovata. Front Plant Sci. 2024; 15: 1354249. doi:10.3389/fpls.2024.1354249. [Google Scholar] [CrossRef]
98. Wang SM , Wang YS , Cheng H . Comparative transcriptomics and metabolomics analyses of Avicennia marina and Kandelia obovata under chilling stress during seedling stage. Int J Mol Sci. 2023; 24( 23): 16989. doi:10.3390/ijms242316989. [Google Scholar] [CrossRef]
99. Mascher M , Jayakodi M , Shim H , Stein N . Promises and challenges of crop translational genomics. Nature. 2024; 636( 8043): 585– 93. doi:10.1038/s41586-024-07713-5. [Google Scholar] [CrossRef]
100. Li D , Quan C , Song Z , Li X , Yu G , Li C , et al. High-throughput plant phenotyping platform (HT3P) as a novel tool for estimating agronomic traits from the lab to the field. Front Bioeng Biotechnol. 2020; 8: 623705. doi:10.3389/fbioe.2020.623705. [Google Scholar] [CrossRef]
101. Poorter H , Hummel GM , Nagel KA , Fiorani F , von Gillhaussen P , Virnich O , et al. Pitfalls and potential of high-throughput plant phenotyping platforms. Front Plant Sci. 2023; 14: 1233794. doi:10.3389/fpls.2023.1233794. [Google Scholar] [CrossRef]
102. Smith DT , Potgieter AB , Chapman SC . Scaling up high-throughput phenotyping for abiotic stress selection in the field. Theor Appl Genet. 2021; 134( 6): 1845– 66. doi:10.1007/s00122-021-03864-5. [Google Scholar] [CrossRef]
103. Gill T , Gill SK , Saini DK , Chopra Y , de Koff JP , Sandhu KS . A comprehensive review of high throughput phenotyping and machine learning for plant stress phenotyping. Phenomics. 2022; 2( 3): 156– 83. doi:10.1007/s43657-022-00048-z. [Google Scholar] [CrossRef]
104. Li G , An L , Yang W , Yang L , Wei T , Shi J , et al. Integrated biotechnological and AI innovations for crop improvement. Nature. 2025; 643( 8073): 925– 37. doi:10.1038/s41586-025-09122-8. [Google Scholar] [CrossRef]
105. Tsega A , Mullualem D . Machine learning for multi-omics data integration in crop improvement: a systematic review. BMC Bioinform. 2026; 27( 1): 81. doi:10.1186/s12859-026-06438-8. [Google Scholar] [CrossRef]
106. Grones C , Eekhout T , Shi D , Neumann M , Berg LS , Ke Y , et al. Best practices for the execution, analysis, and data storage of plant single-cell/nucleus transcriptomics. Plant Cell. 2024; 36( 4): 812– 28. doi:10.1093/plcell/koae003. [Google Scholar] [CrossRef]
107. Zhu T , Li T , Lü P , Li C . Single-cell omics in plant biology: mechanistic insights and applications for crop improvement. Adv Biotechnol. 2025; 3( 3): 20. doi:10.1007/s44307-025-00074-8. [Google Scholar] [CrossRef]
108. Moskal M . Single-cell and spatial transcriptomics in plant science. Int J Mol Sci. 2025; 26( 24): 11819. doi:10.3390/ijms262411819. [Google Scholar] [CrossRef]
109. Hu Y , Dash L , May G , Sardesai N , Deschamps S . Harnessing single-cell and spatial transcriptomics for crop improvement. Plants. 2024; 13( 24): 3476. doi:10.3390/plants13243476. [Google Scholar] [CrossRef]
110. Seyfferth C , Renema J , Wendrich JR , Eekhout T , Seurinck R , Vandamme N , et al. Advances and opportunities in single-cell transcriptomics for plant research. Annu Rev Plant Biol. 2021; 72: 847– 66. doi:10.1146/annurev-arplant-081720-010120. [Google Scholar] [CrossRef]
111. Wang H , Cimen E , Singh N , Buckler E . Deep learning for plant genomics and crop improvement. Curr Opin Plant Biol. 2020; 54: 34– 41. doi:10.1016/j.pbi.2019.12.010. [Google Scholar] [CrossRef]
112. Zhang Y , Malzahn AA , Sretenovic S , Qi Y . The emerging and uncultivated potential of CRISPR technology in plant science. Nat Plants. 2019; 5( 8): 778– 94. doi:10.1038/s41477-019-0461-5. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools