
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Genetic Analysis of Variants of the MYH6 Gene Promoter in Congenital Atrial Septal Defects
1
Clinical School of Cardiovascular Disease, Department of Cardiovascular Surgery, TEDA International Cardiovascular Hospital,
Tianjin Medical University, Tianjin, 300457, China
2
The Institute of Cardiovascular Diseases & Department of Cardiovascular Surgery, TEDA International Cardiovascular Hospital,
Tianjin University & Chinese Academy of Medical Sciences, Tianjin, 300457, China
* Corresponding Authors: Guo-Wei He. Email: ; or
Congenital Heart Disease 2023, 18(1), 7-21. https://doi.org/10.32604/chd.2022.025451
Received 14 July 2022; Accepted 16 August 2022; Issue published 09 January 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Atrial septal defect (ASD) is one of the common congenital heart diseases. The MYH6 gene has a critical role in cardiac development but the role of MYH6 promoter variants in patients with ASD has not been explored. Methods: In 613 subjects including 320 ASD patients, we investigated the MYH6 gene promoter variants and verified the effect on gene expression by using cellular functional experiments and bioinformatics analysis. Results: Eleven variants were identified in the MYH6 gene promoter, of which four variants were found only in ASD patients, and two variants (g.3434G>C and g.4524C>T) were identified for the first time. Cellular functional experiments indicated that all four variants reduced the transcriptional activity of the MYH6 gene promoter (p < 0.05). Subsequent analysis through the JASPAR (A database of transcription factor binding profiles) suggests that these variants may alter transcription factor binding sites, which may in turn lead to changes in myocardin subunit expression and ASD formation. Conclusions: Our study for the first time focuses on variants in the promoter region of the MYH6 gene in Chinese patients with ASD and the discovered variants have functional significance. The study provides new insights in the role of the MYH6 gene promoter region to better understand the genetic basis of ASD formation and facilitates clinical diagnosis.Graphic Abstract
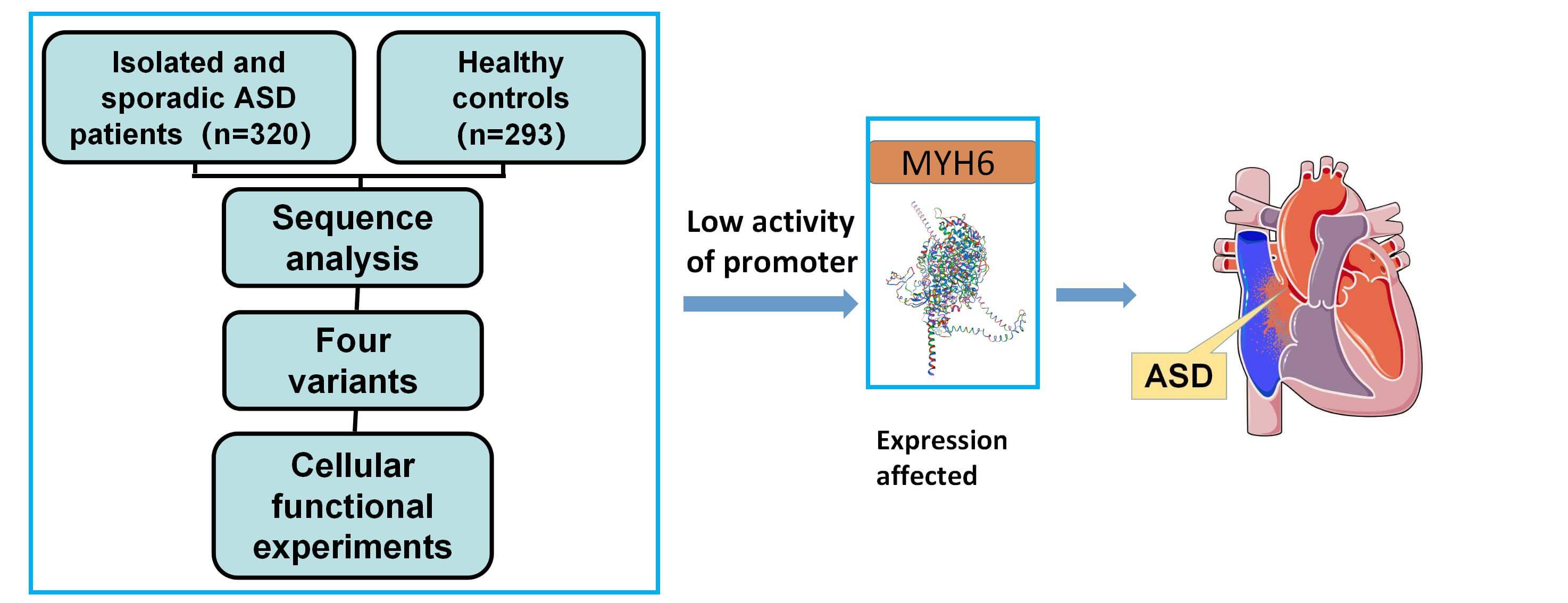
Keywords
Congenital defects pose a serious threat to the health of newborns, the most common of which is congenital heart disease (CHD) (about one-third of the total) [1]. CHD affects approximately 1 in 100 births and is a major risk factor for termination of pregnancy and fetal death [1,2]. Although the mortality rate of CHD has declined with major improvements in medical treatments, CHD remains the leading cause of death in the first year of life, affecting millions of newborns each year. In fact, CHD causes morbidity and hospitalization in children, increases the health care burden, and often requires lifelong follow-up, making it a worldwide public health challenge [3]. CHDs include isolated occurrences and multiple defects occurring simultaneously, and their severity is determined by the location and combination of associated anatomical defects. The causes of these malformations are usually considered multifactorial, in which genetic variation is one of the key causes [4]. It is estimated that the presence of nearly 30% of confirmed cases is due to genetic as well as maternal factors, some of which coincide with other disorders, such as Down syndrome [4–6]. The same type of CHD may be caused by alterations in different genes, and alterations in individual genes may result in different CHDs [7,8]. After decades of research, a large number of genes have now been identified in association with the development of CHD, including structural proteins, transcription factors, and cell signaling molecules that are important for heart development [9].
Atrial septal defect (ASD) is considered one of the most common CHDs, and its incidence is about 10%–20% of all CHDs [2,10]. With the improvement of echocardiographic recognition of clinically asymptomatic ASDs, there is a tendency to increase the detection rate of its occurrence [11,12]. Many isolated ASDs do not produce severe symptoms, which makes many ASDs difficult to detect in childhood, resulting in many ASD patients to receive treatment in adulthood. The surgical procedure for adults with ASD has a higher mortality rate and a higher incidence of complications such as right heart dilation, arrhythmias, pulmonary hypertension, and complete atrioventricular block [6]. Over the past two decades, the treatment of ASD has evolved considerably, not only through surgery, but also through catheter closure, but the cost of treatment is still a huge burden [13]. This prompts us to raise awareness of ASD and reduce its occurrence at the root. Genetic variation has been shown to be an important cause of ASD [14], but many of the genes that have an impact on the formation of ASD remain undiscovered, and thus there is still much work needed to be done.
The MYH6 (OMIM: 160710) gene located on chromosome 14q11.2 encodes the α subunit of the heavy chain of cardiac myosin [15]. It is involved in the formation of the heart since the beginning of embryonic development and is one of the most important subunits that constitute the composition of cardiac myosin [16]. The variants of MYH6 have important implications for many types of CHD and cardiomyopathy, including Shone complex, aortic constriction, hypoplastic left heart syndrome, hypertrophic cardiomyopathy, and dilated cardiomyopathy [15–18]. MYH6 is abundantly expressed in atrial muscle, and mutations in several exonic regions of the MYH6 gene have been found to contribute to the formation of ASD [19,20]. Further, in the study of genetic effects on CHD, studies have been focused on variants in non-coding regions [21]. The promoter in the non-coding region is one of the important factors of gene transcription. As we previously reported, variants of the promoter region of CITED2 [22] and MEF2C [23] resulted in the formation of CHD. However, no studies on the effect of variants in the MYH6 promoter region on ASD have been reported.
Based on the above, we hypothesized that variants in the MYH6 promoter may affect MYH6 gene expression and may be involved in the formation of ASD. To test this hypothesis, we designed the present study to investigate the relationship between MYH6 gene promoter variants and ASD formation by performing genetic and functional analysis of DNA sequences of MYH6 promoter region in 613 Chinese subjects (320 patients with ASD and 293 healthy controls).
A total of 613 subjects were enrolled in this study, including 320 isolated simple ASD patients and 293 healthy controls. All patients underwent ASD repair in TEDA International Cardiovascular Hospital, Tianjin, China. Patients with a family history of CHD or concomitant genetic disorders of other diseases were excluded. The healthy controls were clinically confirmed free of CHD by Doppler echocardiography and other clinical examinations, and had no family history of relevant genetic diseases. This work was performed in accordance with the principles of the Declaration of Helsinki and was approved by the Ethics Committee of the TEDA International Cardiovascular Hospital. Written consent forms were informed and signed by the parents or guardians of the participants. The flow chart of this study is shown in Fig. 1.
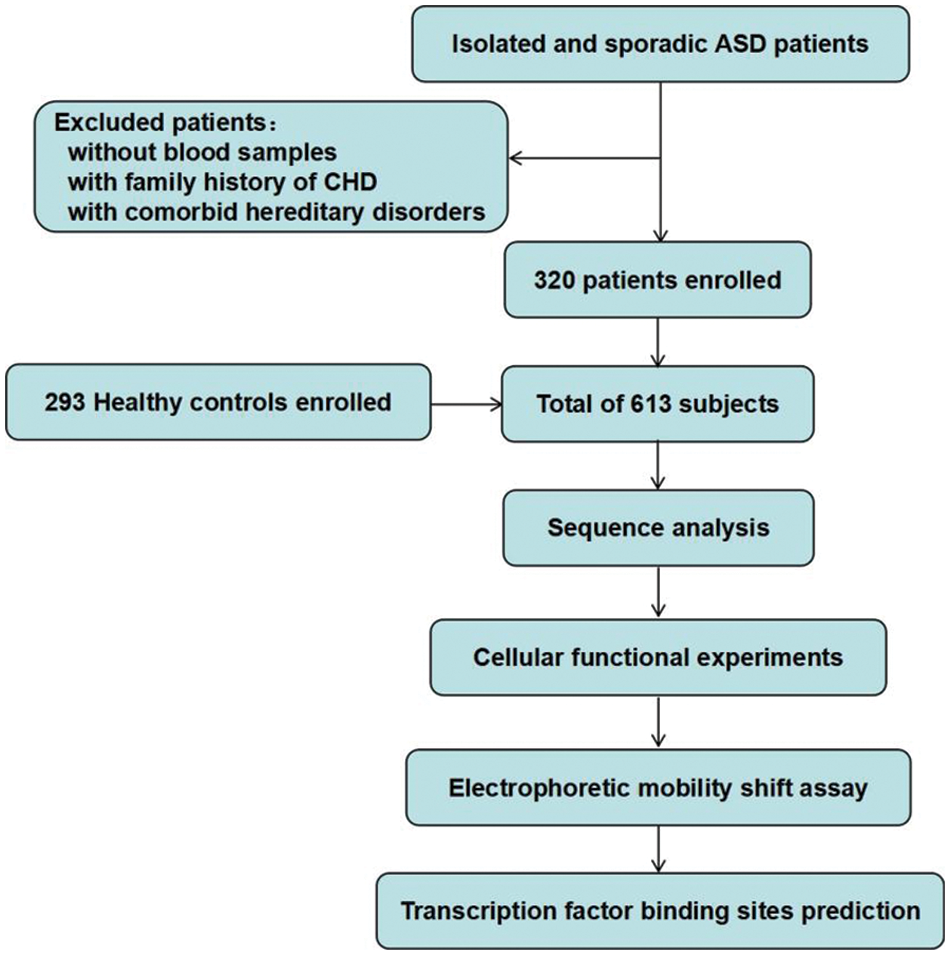
Figure 1: Study flow chart. A total of 613 subjects were recruited. DNA was extracted from blood samples. Sequence analysis and cellular function experiments were performed to identify the variants. ASD, atrial septal defect; CHD, congenital heart defect
Genomic DNA was extracted using a DNA extraction kit, and 50 μl of DNA was extracted from each sample and stored at −80°C. The reference sequence of MYH6 gene promoter was obtained from GenBank database (NCBI, NG_023444.1). The design of PCR primers was based on the reference sequence (NCBI: NG_016169.1) to cover specific MYH6 promoter sequence (1638 bp, transcription start site −1791 to −154 bp). The primer sequences are listed in Table 1. The amplification conditions were: denaturation at 95°C for 5 min, followed by 35 cycles (denaturation at 95°C for 30 s, annealing at 60°C for 30 s, extension at 72°C for 105 s), and finally extension at 72°C for 10 min. The amplified DNA products were collected for bidirectional Sanger sequencing, and the DNA sequences of patients were compared with controls to screen variants. The primers for PCR and Sanger sequencing are shown in Table 1.
2.3 Plasmid Construction, Cell Culture and Transfection
To determine whether the variants affect the activity of promoter, we inserted the MYH6 gene promoter region, with or without variants, into the KpnI and SacI sites of pGL6 basic (firefly luciferase reporter vector). The PCR primers with expression vectors for KpnI and SacI sites are shown in Table 1. Subsequently, the expression-loaded plasmids with the target genes were transformed into E. coli receptor cells, transferred into the medium, and incubated for 16 h at 37°C and 200 rpm for propagation. According to the experimental protocol, we extracted plasmids from E. coli using a plasmid miniprep kit (Beyotime Biotechnology, Shanghai, China). The pGL6 basic-MYH6 promoter plasmids were co-transfected with pRL-SV40 (Renilla luciferase reporter plasmid) at a 5:1 ratio into HEK-293 cells and cultured for 24–48 h in six-well plates. The pRL-SV40 was used as the internal control. The empty pGL6-basic was used as a negative control. The cells were cultured in minimum essential medium without antibiotics added with 10% fetal bovine serum for 24–48 h (1 × 106 cells per 2.5 ng plasmid DNA transfection). This was followed by a dual luciferase activity assay.
2.4 Analysis by Dual Luciferase Reporter Gene Assay
After transfection is completed, the cells were collected and lysed sufficiently to retain the lysate, which was dripped into a 96-well plate. Firefly and Renilla luciferase activities of the cell lysates were measured by sequentially adding reagents from the Dual Luciferase Reporter Assay Kit (Beyotime Biotechnology, Shanghai, China) according to the instructions. The empty vector pGL6-basic plasmid and blank vector were used as negative controls, and pRL-SV40 was used as an internal reference control to correct variations in transfection efficiency. All experiments were independently replicated four times.
2.5 Preparation of Nuclear Extracts and Determination of Electrophoretic Mobility Shift Assay (EMSA)
Transcription factors (TFs) regulate the target gene expression by recognizing TF binding sites (TFBS) located in cis-regulatory regions of target genes (promoters, enhancers) and interact with DNA in a specific manner [24]. EMSA assay was used to detect the effect of variants identified in MYH6 promoter on potential transcription factors in the present study. First, nuclear proteins were extracted from HEK-293 cells and HL-1 cells using Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime Biotechnology, Shanghai, China). Subsequently, nuclear protein concentrations were measured using the Enhanced BCA Protein Assay Kit (Beyotime Biotechnology, Shanghai, China) after storage at −80°C. Biotinylated double-stranded oligonucleotide probes were prepared based on the MYH6 gene promoter with or without variants, annealed at 95°C for one hour and prepared for further use.
EMSA chemiluminescence kit (Beyotime Biotechnology, Shanghai, China) was used with equal amounts of probe (0.25 pM) and nuclear extract (2.0 mg) in each tube, according to the manufacturer’s protocol. Sequences of probes are shown in Table 1. The experimental procedure for the preparation of probe, nuclear protein, and EMSA is shown in Figs. 2A and 2B.
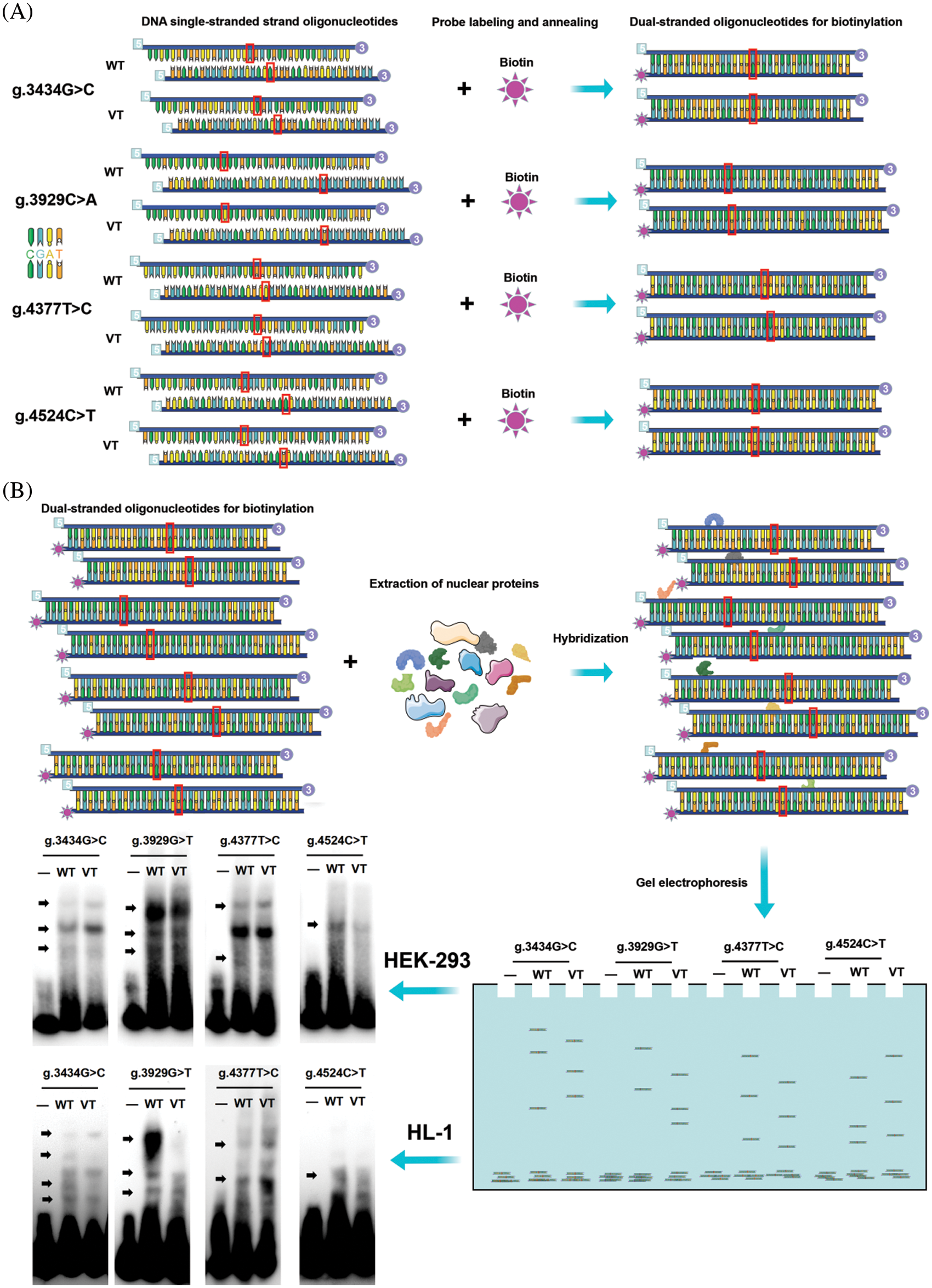
Figure 2: EMSA procedure. (A) Preparation of biotin double-stranded oligonucleotide probes. The designed single-stranded nucleotide sequences were labeled with biotin. The complementary sequences were annealed and hybridized. (B) Biotin probes were crossed to the wild-type and variant-type of the four variants respectively, nuclear proteins were extracted from HEK-293 cells and HL-1 cells, and then subjected to EMSA separately. Four variants (g.3434G>C, g.3929C>A, g.4377T>C and g.4524C>T) were seen to create or abolish the binding of TFs in HEK-293 cells. EMSA, electrophoretic mobility shift assay; WT, wild-type; VT, variant-type; TF, transcription factor
In view of the importance of TF-promoter interactions in the study of gene expression regulation, there are several computational approaches to model and predict TFBS. The JASPAR database (https://jaspar.genereg.net/) is a widely used open access database that allows prediction of TFBS on promoter region from multiple data modules [25]. To further investigate whether variants affected original TFBS in MYH6 gene promoter, we used JASPAR to go through the list of all potentially affected binding sites for the identified variants, with the relative spectrum score threshold set at 85%. The filtered differential TFBS were subjected to comparative analysis. The wild-type and variant-type nucleotide sequences are shown in Table 1.
Quantitative data were expressed as mean ± standard deviation and compared using one-way ANOVA. All statistical analyses were performed using SPSS 25.0 software. p < 0.05 was considered statistically significant.
3.1 Variants Identified by DNA Sequencing in Patients with ASD
In the present study, 11 variants were identified by Sanger sequencing. All these variants are summarized in Table 2. Their locations are shown in Fig. 3A and their sequencing chromatograms are shown in Figs. 3B and 3C. Among these 11 variants, 4 variants [g.3434G>C, g.3929C>A (rs933962908), g.4377T>C (rs373958405) and g.4524C>T] were found only in ASD patients, while the other 7 variants were found in both ASD patients and healthy controls (Table 2). Remarkably, two of these four variants (g.3434G>C and g.4524C>T) have not been reported. The allele frequency of the variant g.3929C>A (rs933962908) variant was <0.001 (T = 0.00018) in the NCBI SNP database, and this variant occurred in the same patient who also had the variant g.3434G>C. These four variants were further investigated in cellular function experiments to test the possible pathogenic effect whereas the other seven variants found in both ASD patients and controls were excluded from further studies.
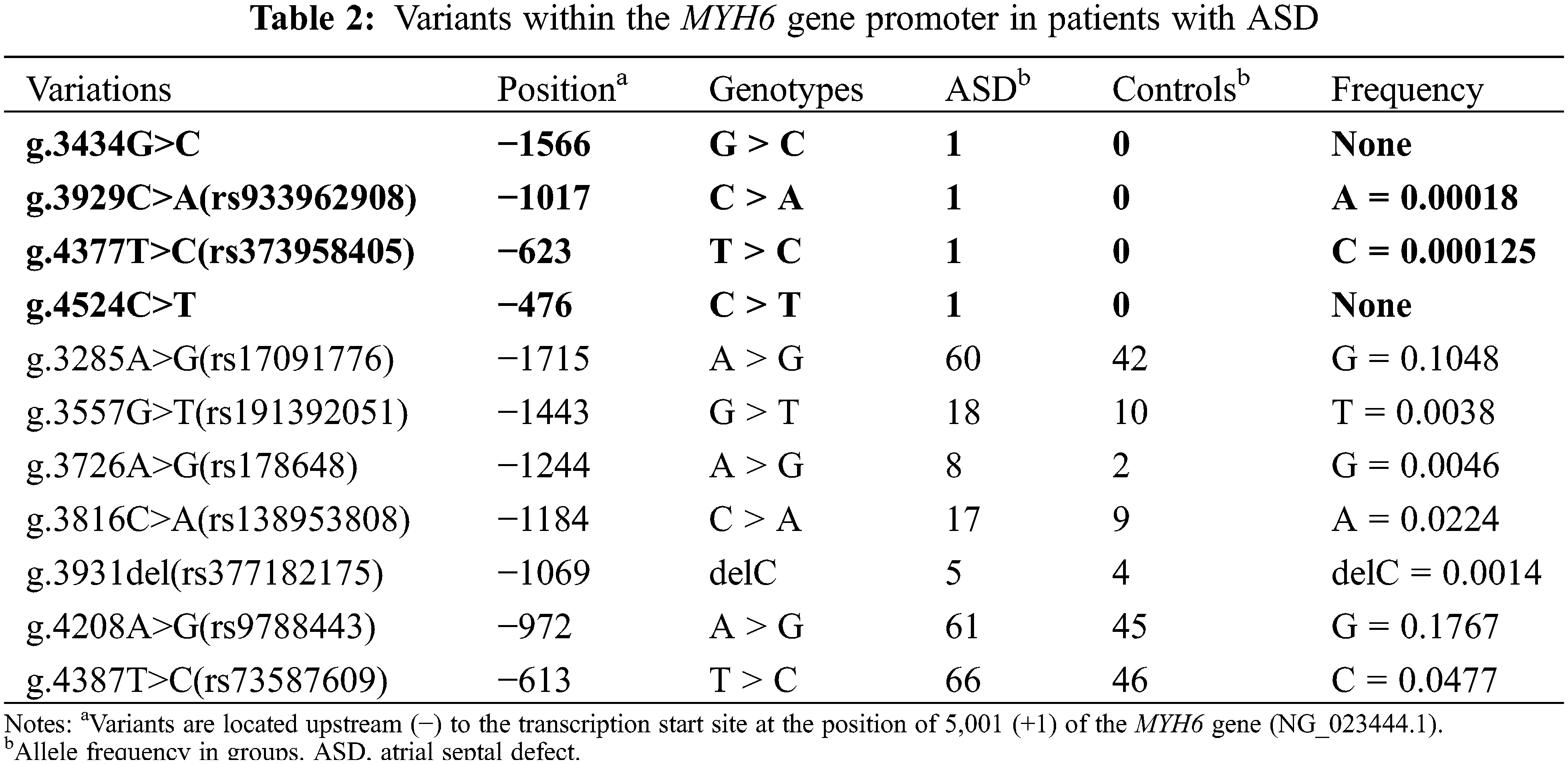
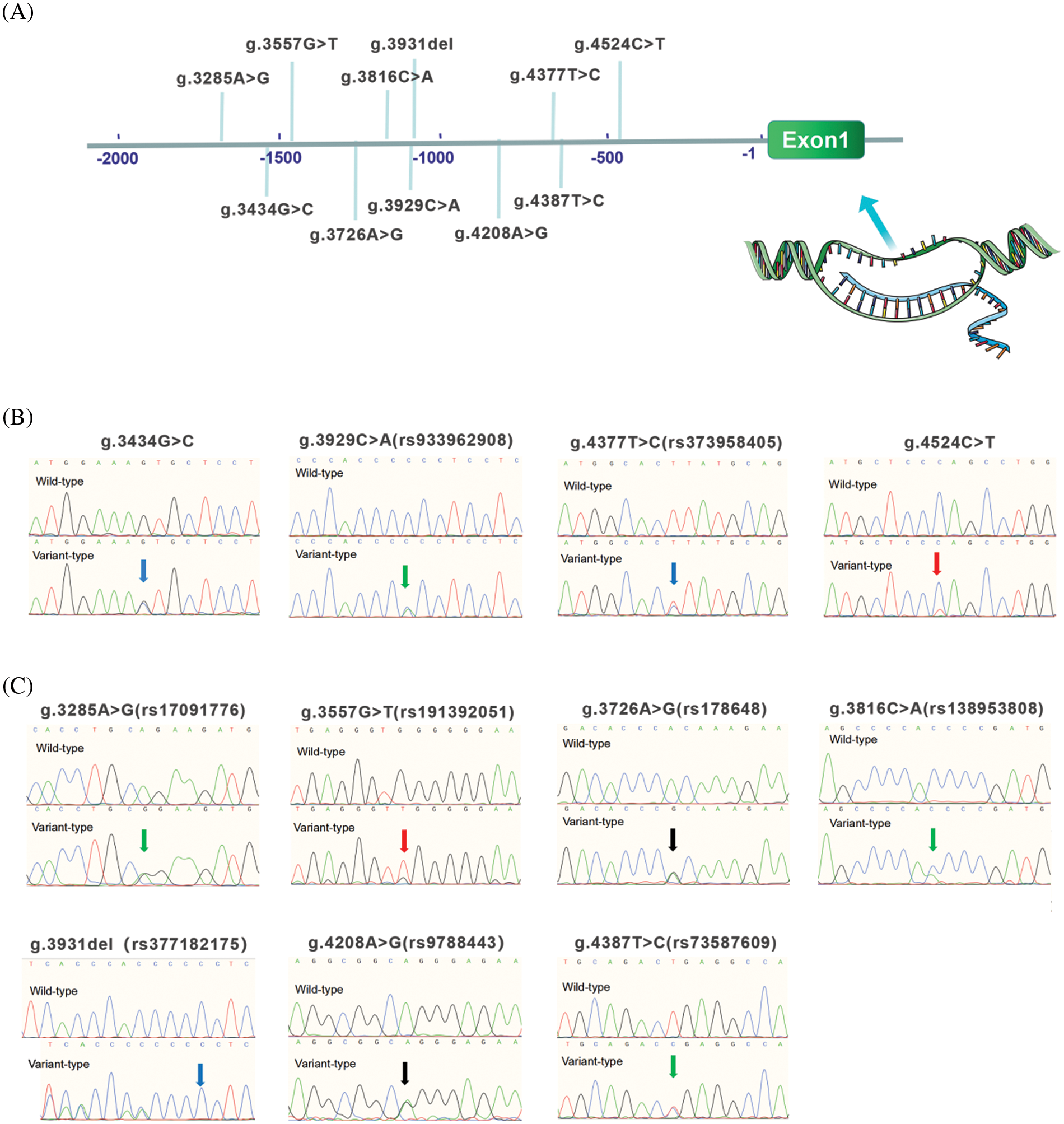
Figure 3: Locations and sequencing chromatograms of MYH6 gene promoter variants. (A) Genetic variants are named according to the genomic DNA sequence of the human MYH6 gene (Genbank accession number NG_023444.1). The transcription start site is at position 5,001 in the first exon. (B) Sequencing chromatograms of 4 variants found only in ASD patients. Top panels show wild-type and bottom panels show variant-type, marked with arrows. (C) Sequencing chromatograms of 7 variants found in both ASD patients and controls. Top panels show wild-type and bottom panels show variant-type, marked with arrows. ASD, atrial septal defect
3.2 Regulatory Variants Alter the Activity of the MYH6 Gene Promoter
The wild-type and variant-type MYH6 gene promoters were cloned into the firefly luciferase reporter plasmid (pGL6-basic) reporter vector, and expression vectors were constructed, including blank, empty pGL6-basic (negative control), pGL6-WT (wild-type MYH6 gene promoter), pGL6-3434C (g.3434G>C), pGL6-3929T (g.3929C>A), pGL6-4377C (g.4377T>C) and pGL6-4524T (g.4524C>T). These constructed vectors were then transfected into HEK-293 cells together with pRL-SV40 and dual luciferase activity was measured. The transcriptional activities of the blank control and negative control groups were much lower than the other five groups, approaching 0 (Fig. 4A). The four variant gene promoters, found only in ASD patients, significantly reduced gene promoter expression activity and luciferase expression [g.3434G>C and g.4377T>C (rs373958405) (p < 0.05); g.4524C>T (p < 0.01); g.3929C>A (rs933962908) (p < 0.001)] (Fig. 4A). Fig. 4B shows the color Doppler echocardiograms of patients with these four variants, indicating a large ASD in each of these three patients.
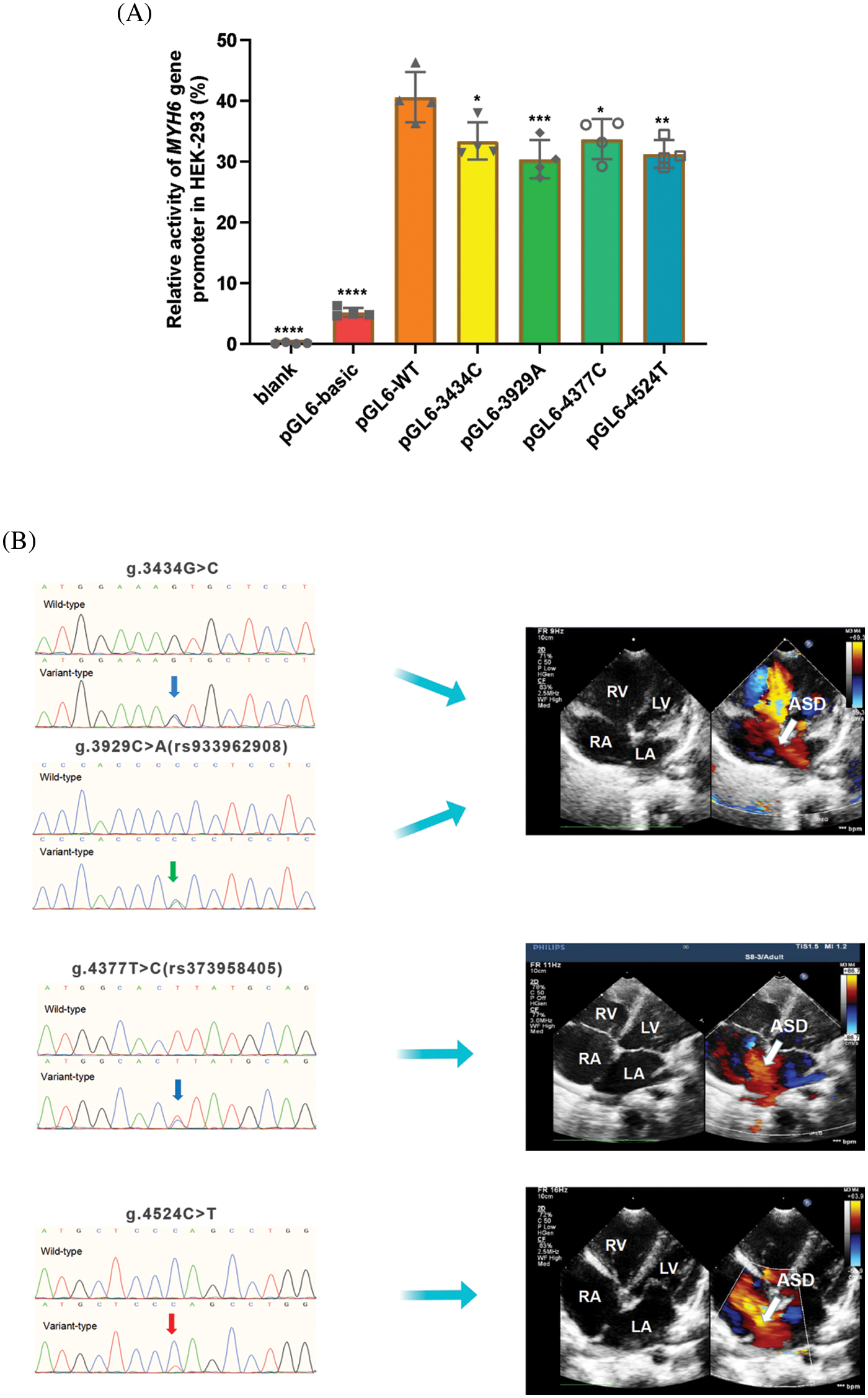
Figure 4: Results of cellular function tests. (A) Relative transcriptional activity of wild-type and variants of MYH6 gene promoters in HEK-293. Quantitative data are expressed as mean ± SD and are based on four independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). (B) Color Doppler echocardiography shows ASD (arrow) in patients with variants of g.3434G>C, g.3929C>A (rs933962908), g.4377T>C (rs373958405) and g.4524C>T. LA, left atrium; RA, right atrium; RV, right ventricle; LV, left ventricle; ASD, atrial septal defect
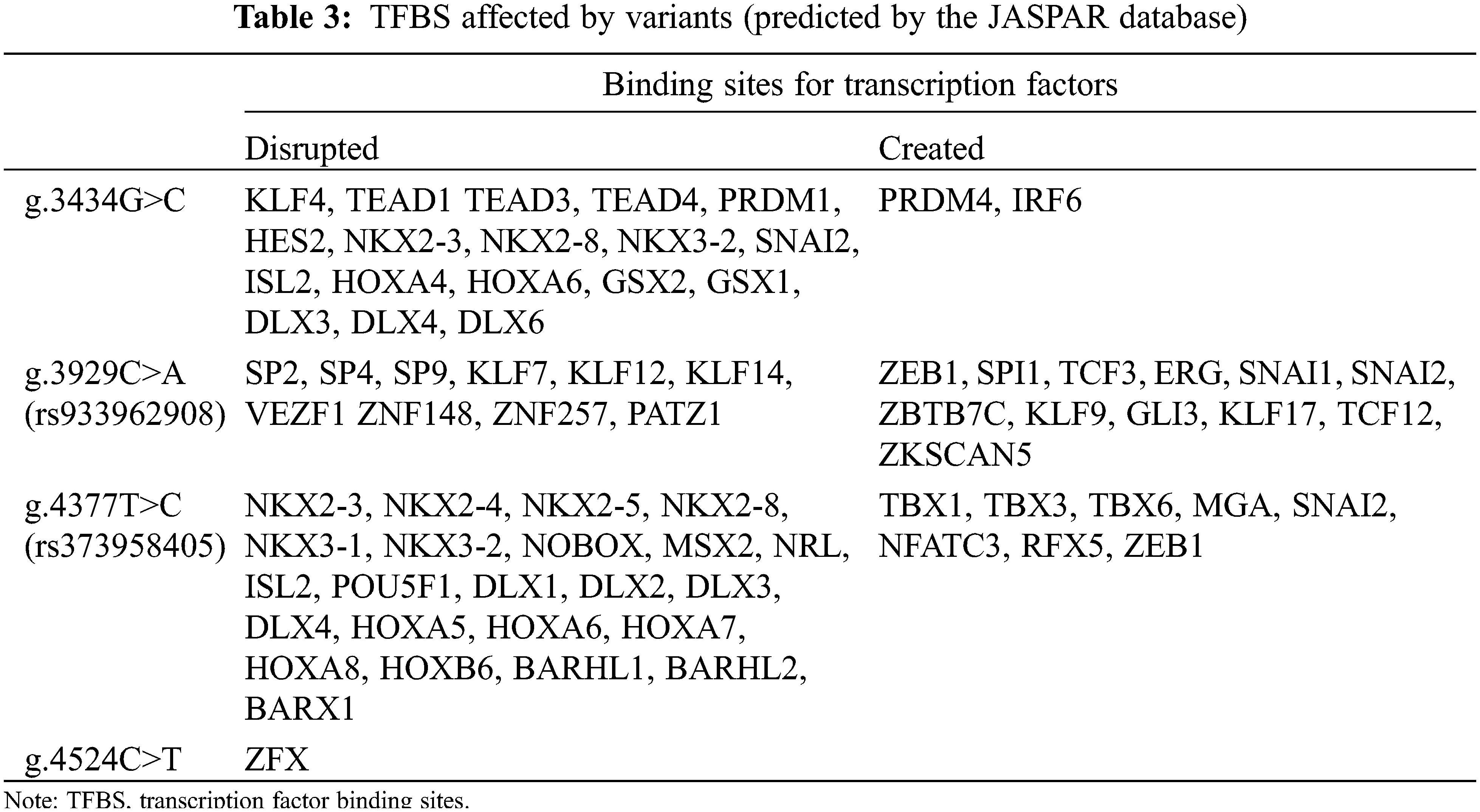
We performed EMSA assays using wild-type or variant biotin-labeled double-stranded probes, examining the four variants found only in ASD patients. The biotinylated oligonucleotides of EMSA are shown in Table 1. As shown in Fig. 2B, the arrows indicate bands with different levels of lightness and darkness, indicating that different numbers of TFs are bound at that location. When the bands are lighter, it means that there were fewer TFs bound on the location. In contrast, when the bands are darker, it means that there were more TFs bound on the location. Table 3 shows the TFs bound at the variant site, either disrupted or created by the variants, predicted by JASPAR database (see below). Taken together, these results demonstrated that the ability of the probe with four variants [g.3434G>C, g.3929C>A (rs933962908), g.4377T>C (rs373958405), and g.4524C>T] to bind to TFs in HEK-293 cells and HL-1 cells were either reduced or increased compared to the wild type.
3.4 The TFBS Affected by Regulatory Variants
Based on the JASPAR core TF database, we predicted potential TFBS that could be disrupted or generated by variants in the MYH6 gene promoter. The four variants were identified with a total of 43 potential new TFBS and 22 reduced TFBS. In reduced TFBS, multiple transcription factors play important roles in cardiogenesis, such as NKX2-5, KLF4, TEAD1, DLX6, SP2, HOXA5, and MSX2 [26–32], as shown in Table 3. Based on the results and the above analyses, as well as previous studies, we established a schema describing the possible effects of MYH6 gene promoter region variants on ASD formation, as shown in Fig. 5.
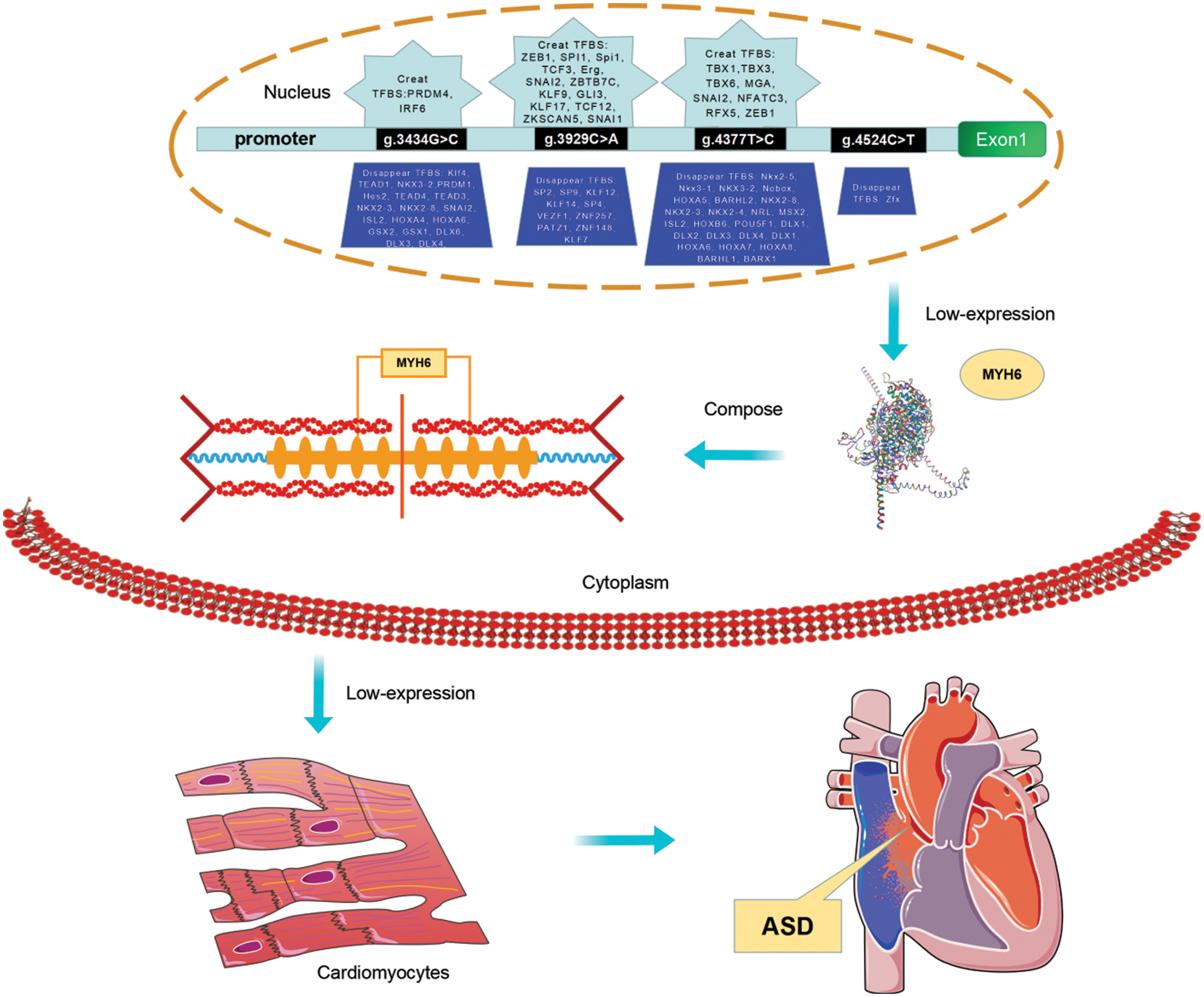
Figure 5: The schema describing the role of variants of MYH6 gene promoter. The schema illustrates the role of variants in the promoter region of the MYH6 gene identified from the present study and relates to the analysis of the JASPAR database and the results of previous studies. To summarize, the variants in the MYH6 gene promoter identified in the study may alter the TFBS cluster, which leads to altered gene expression and consequently affects the formation of cardiac myosin heavy chain. And this is directly related to the development of ASD. In addition, variation in the MYH6 promoter affects NKX2-5, KLF4, TEAD1, DLX6, SP2, HOXA5, and MSX2, and the binding of transcription factors such as may lead to the formation of ASD. TF, transcription factor; TFBS, transcription factor binding sites; ASD, atrial septal defect
This study for the first time found in Chinese ASD patients that (1) there are a total of 11 variants of MYH6 gene promoter identified in the 613 patients enrolled in the study, of which 4 variants [g.3434G>C, g.3929C>A (rs933962908), g.4377T>C (rs373958405) and g.4524C>T] are unique to ASD patients. Interestingly, two of these four variants (g.3434G>C and g.4524C>T) are identified for the first time. Besides, variants g.3929C>A and g.3434G>C occurred in the same patient; (2) all four variants were found to result in a significant reduction in promoter transcriptional activity by a dual luciferase activity cell function assay (p < 0.05); and (3) by EMSA and bioinformatics analyses these four variants contributed to the alteration of TFBS and the reduced transcriptional activity of MYH6 gene is potentially involved in the formation of ASD.
Myosin is one of the essential components of the myocardium and its heavy chain is mainly encoded by MYH6 and MYH7 genes in myocardium. MYH6 gene can encode its own protein and affect the expression of MYH7 gene [33]. Experimental animal studies have suggested that some mutations in MYH6 gene are lethal, resulting in the inability to form myofibers properly during myocardial development and affecting the formation of myocardial septa further [34].
As a key gene in cardiac development, it has been shown that ASD can be developed by mutations in the coding region of the MYH6 gene in humans [20,35] and by MYH6 knockdown in chicken embryos [34]. However, whether MYH6 gene variants in the promoter region affect the development of the atrial septum has not been reported. In our study population (n = 613), four variants were identified in the MYH6 gene promoter region in three ASD patients with a frequency of 0.94% (3/320) in ASD patients and 0.49% (3/613) in the total study population.
The promoter of the MYH6 gene has been used to induce the desired phenotype in mouse studies [36], indicating that the MYH6 promoter plays an irreplaceable role in the formation of the heart. In this study, we measured the transcriptional activity of the variant promoter regions by means of dual fluorescein reporter analysis. The present study demonstrates that the transcriptional activity of the promoter at the variant region of MYH6 was decreased. This would most likely lead to low expression of the MYH6 gene and has significant pathological role in the formation of the atrial septum, as mentioned above [20,34,35].
Furthermore, the present study demonstrated that all four variants [g.3434G>C, g.3929C>A (rs933962908), g.4377T>C (rs373958405) and g.4524C>T] found only in ASD patients had altered binding affinity for the extracted nuclear proteins. This result is in accordance with previous studies indicating that variations in the promoter region lead to altered binding to TFs and contribute to a number of diseases [22,37].
The promoter can regulate transcriptional activity by binding to TFs [38]. According to the JASPAR database prediction analysis, the four variants [g.3434G>C, g.3929C>A (rs933962908), g.4377T>C (rs373958405), and g.4524C>T] found in the present study in the ASD patients have been predicted to have a large number of TFBS disrupted. In contrast, only a small amount of TFBS was produced in the variants. While in the analysis of these TFBS, variant g.4377T>C (rs373958405) has an important TFBS NKX2-5 disrupted that is noteworthy. NKX2-5 has been demonstrated in many studies to play an important role in cardiac development [39,40], and has an essential relationship with the formation of the atria [29,41]. In fact, it has been shown that NKX2-5 promotes the expression of MYH6 gene [42]. In addition, the other three variants also disrupted a number of TFBS with key roles in cardiovascular development and repair, such as KLF4, TEAD1, DLX6, SP2, HOXA5, and MSX2 [26–28,30–32]. There are also other disrupted TFBS whose role in cardiovascular development has not been clarified (Table 3).
Fig. 5 shows a diagram that describes the role of variants in the MYH6 gene promoter found in the present study. The low expression of MYH6 gene promoter activity caused by the variants may contribute to the formation of ASD. In addition, variants in the MYH6 promoter region may interfere with the formation of transcriptional regulatory complexes with related TFs, such as NKX2-5, KLF4, SP2, HOXA5, and MSX2, affecting the development of the atrial septum and resulting in the formation of ASD.
We realize that this study has some limitations. Whether the effect of the variants found in this study directly leads to the development of ASD needs further studies to confirm. As we described in the Fig. 5, there are other genes that may interact with the MYH6 in the development of the heart. The results from the cellular experiments may be further validated in animal models. Further, in a larger cohort of ASD patients, it is possible to discover more variants in the MYH6 promoter that possibly have pathogenic role in the development of ASD. These will be our future direction of study.
In summary, our study identified 11 variants in the promoter region of the MYH6 gene in Chinese patients with ASD. Four of them were found only in ASD patients, and two of them were identified for the first time. In addition, cell function experiments, EMSA and bioinformatics analysis showed that these four variants significantly altered MYH6 gene expression and affected the binding of TFs, which may facilitate the development of ASD. Therefore, the present study provides insights into further understanding of the role of gene promoter region in ASD formation and into the mechanisms of ASD formation.
Acknowledgement: We thank the patients and their family members for cooperation. The assistance of Nursing staff at the Division of Pediatric Cardiac Surgery, Department of Cardiovascular Surgery is gratefully acknowledged.
Authorship: G.W.H. conceptualized the project. G.W.H. and H.X.C., designed the study. G.W.H. obtained the funding for this project. J.Y.Z., H.X.C. performed experiments. J.Y.Z., H.X.C., and G.W.H. analyzed experimental data. J.Y.Z., H.X.C., Z.G.L., and G.W.H. collected the human blood samples. Q.Y. discussed the findings and participated in the project. J.Y.Z., and G.W.H. wrote the manuscript. G.W.H. supervised the whole project.
Ethics Approval: This study involving human participants was reviewed and approved by the ethics committee of TEDA International Cardiovascular Hospital, China (No. 0715-4, 2021, 02 August 2021).
Availability of Data and Materials: The data underlying this article will be shared on reasonable request to the corresponding author.
Funding Statement: This work was funded by the National Natural Science Foundation of China [82170353 & 81870288]; the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences [2020-PT310-007]; Tianjin Municipal and Binhai New Area Health Commissions [KJ20071 & 2019BWKY010]; Tianjin Science and Technology Project [18PTZWHZ00060]; TEDA International Cardiovascular Hospital [2021-TD-006 & 2021-ZX-002 & 2019-TD-013]; and Tianjin Key Medical Discipline (Specialty) Construction Project [TJYXZDXK-019A].
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Houyel, L., Meilhac, S. M. (2021). Heart development and congenital structural heart defects. Annual Review of Genomics and Human Genetics, 22(1), 257–284. DOI 10.1146/annurev-genom-083118-015012. [Google Scholar] [CrossRef]
2. Zhao, Q. M., Liu, F., Wu, L., Ma, X. J., Niu, C. et al. (2019). Prevalence of congenital heart disease at live birth in China. Journal of Pediatrics, 204(Suppl 2), 53–58. DOI 10.1016/j.jpeds.2018.08.040. [Google Scholar] [CrossRef]
3. Liu, Y., Chen, S., Zühlke, L., Black, G. C., Choy, M. K. et al. (2019). Global birth prevalence of congenital heart defects 1970–2017: Updated systematic review and meta-analysis of 260 studies. International Journal of Epidemiology, 48(2), 455–463. DOI 10.1093/ije/dyz009. [Google Scholar] [CrossRef]
4. Diz, O. M., Toro, R., Cesar, S., Gomez, O., Sarquella-Brugada, G. et al. (2021). Personalized genetic diagnosis of congenital feart defects in newborns. Journal of Personalized Medicine, 11(6), 562. DOI 10.3390/jpm11060562. [Google Scholar] [CrossRef]
5. Reamon-Buettner, S. M., Borlak, J. (2004). Somatic NKX2-5 mutations as a novel mechanism of disease in complex congenital heart disease. Journal of Medical Genetics, 41(9), 684–690. DOI 10.1136/jmg.2003.017483. [Google Scholar] [CrossRef]
6. Webb, G., Gatzoulis, M. A. (2006). Atrial septal defects in the adult: Recent progress and overview. Circulation, 114(15), 1645–1653. DOI 10.1161/CIRCULATIONAHA.105.592055. [Google Scholar] [CrossRef]
7. Nees, S. N., Chung, W. K. (2020). The genetics of isolated congenital heart disease. American Journal of Medical Genetics Part C—Seminars in Medical Genetics, 184(1), 97–106. DOI 10.1002/ajmg.c.31763. [Google Scholar] [CrossRef]
8. Van Nisselrooij, A., Lugthart, M. A., Clur, S. A., Linskens, I. H., Pajkrt, E. et al. (2020). The prevalence of genetic diagnoses in fetuses with severe congenital heart defects. Genetics in Medicine, 22(7), 1206–1214. DOI 10.1038/s41436-020-0791-8. [Google Scholar] [CrossRef]
9. Williams, K., Carson, J., Lo, C. (2019). Genetics of congenital heart disease. Biomolecules, 9(12), 879. DOI 10.3390/biom9120879. [Google Scholar] [CrossRef]
10. Geva, T., Martins, J. D., Wald, R. M. (2014). Atrial septal defects. Lancet, 383(9932), 1921–1932. DOI 10.1016/S0140-6736(13)62145-5. [Google Scholar] [CrossRef]
11. Lang, R. M., Mor-Avi, V., Sugeng, L., Nieman, P. S., Sahn, D. J. (2006). Three-dimensional echocardiography: The benefits of the additional dimension. Journal of the American College of Cardiology, 48(10), 2053–2069. DOI 10.1016/j.jacc.2006.07.047. [Google Scholar] [CrossRef]
12. Silvestry, F. E., Kerber, R. E., Brook, M. M., Carroll, J. D., Eberman, K. M. et al. (2009). Echocardiography-guided interventions. Journal of the American Society of Echocardiography, 22(3), 213–317. DOI 10.1016/j.echo.2008.12.013. [Google Scholar] [CrossRef]
13. O’Byrne, M. L., Gillespie, M. J., Shinohara, R. T., Dori, Y., Rome, J. J. et al. (2015). Cost comparison of transcatheter and operative closures of ostium secundum atrial septal defects. American Heart Journal, 169(5), 727–735.e2. DOI 10.1016/j.ahj.2015.02.010. [Google Scholar] [CrossRef]
14. LaHaye, S., Corsmeier, D., Basu, M., Bowman, J. L., Fitzgerald-Butt, S. et al. (2016). Utilization of whole exome sequencing to identify causative mutations in familial congenital heart disease. Circulation: Cardiovascular Genetics, 9(4), 320–329. DOI 10.1161/CIRCGENETICS.115.001324. [Google Scholar] [CrossRef]
15. Bjornsson, T., Thorolfsdottir, R. B., Sveinbjornsson, G., Sulem, P., Norddahl, G. L. et al. (2018). A rare missense mutation in MYH6 associates with non-syndromic coarctation of the aorta. European Heart Journal, 39(34), 3243–3249. DOI 10.1093/eurheartj/ehy142. [Google Scholar] [CrossRef]
16. Jiang, J., Wakimoto, H., Seidman, J. G., Seidman, C. E. (2013). Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science, 342(6154), 111–114. DOI 10.1126/science.1236921. [Google Scholar] [CrossRef]
17. Jin, S. C., Homsy, J., Zaidi, S., Lu, Q., Morton, S. et al. (2017). Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nature Genetics, 49(11), 1593–1601. DOI 10.1038/ng.3970. [Google Scholar] [CrossRef]
18. Theis, J. L., Hu, J. J., Sundsbak, R. S., Evans, J. M., Bamlet, W. R. et al. (2021). Genetic association between hypoplastic left heart syndrome and cardiomyopathies. Circulation: Genomic and Precision Medicine, 14(1), e003126. DOI 10.1161/CIRCGEN.120.003126. [Google Scholar] [CrossRef]
19. Ching, Y. H., Ghosh, T. K., Cross, S. J., Packham, E. A., Honeyman, L. et al. (2005). Mutation in myosin heavy chain 6 causes atrial septal defect. Nature Genetics, 37(4), 423–428. DOI 10.1038/ng1526. [Google Scholar] [CrossRef]
20. Matsson, H., Eason, J., Bookwalter, C. S., Klar, J., Gustavsson, P. et al. (2008). Alpha-cardiac actin mutations produce atrial septal defects. Human Molecular Genetics, 17(2), 256–265. DOI 10.1093/hmg/ddm302. [Google Scholar] [CrossRef]
21. Ye, L., Yu, Y., Zhao, Z. A., Zhao, D., Ni, X. et al. (2022). Patient-specific iPSC-derived cardiomyocytes reveal abnormal regulation of FGF16 in a familial atrial septal defect. Cardiovascular Research, 118(3), 859–871. DOI 10.1093/cvr/cvab154. [Google Scholar] [CrossRef]
22. Zheng, S. Q., Chen, H. X., Liu, X. C., Yang, Q., He, G. W. (2021). Genetic analysis of the CITED2 gene promoter in isolated and sporadic congenital ventricular septal defects. Journal of Cellular and Molecular Medicine, 25(4), 2254–2261. DOI 10.1111/jcmm.16218. [Google Scholar] [CrossRef]
23. Zeng, Z. H., Chen, H. X., Liu, X. C., Yang, Q., He, G. W. (2022). Functional significance of novel variants of the MEF2C gene promoter in congenital ventricular septal defects. American Journal of Medical Genetics Part A, 188(8), 2397–2405. DOI 10.1002/ajmg.a.62871. [Google Scholar] [CrossRef]
24. Lambert, S. A., Jolma, A., Campitelli, L. F., Das, P. K., Yin, Y. et al. (2018). The human transcription factors. Cell, 172(4), 650–665. DOI 10.1016/j.cell.2018.01.029. [Google Scholar] [CrossRef]
25. Castro-Mondragon, J. A., Riudavets-Puig, R., Rauluseviciute, I., Lemma, R. B., Turchi, L. et al. (2022). JASPAR 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Research, 50(D1), D165–D173. DOI 10.1093/nar/gkab1113. [Google Scholar] [CrossRef]
26. Atkinson, A. J., Logantha, S. J., Hao, G., Yanni, J., Fedorenko, O. et al. (2013). Functional, anatomical, and molecular investigation of the cardiac conduction system and arrhythmogenic atrioventricular ring tissue in the rat heart. Journal of the American Heart Association, 2(6), e000246. DOI 10.1161/JAHA.113.000246. [Google Scholar] [CrossRef]
27. Azakie, A., Fineman, J. R., He, Y. (2006). Sp3 inhibits Sp1-mediated activation of the cardiac troponin T promoter and is downregulated during pathological cardiac hypertrophy in vivo. American Journal of Physiology-Heart and Circulatory Physiology, 291(2), H600–H611. DOI 10.1152/ajpheart.01305.2005. [Google Scholar] [CrossRef]
28. Gao, Y., Sun, Y., Ercan-Sencicek, A. G., King, J. S., Akerberg, B. N. et al. (2021). YAP/TEAD1 complex is a default repressor of cardiac toll-like receptor genes. International Journal of Molecular Sciences, 22(13), 6649. DOI 10.3390/ijms22136649. [Google Scholar] [CrossRef]
29. McElhinney, D. B., Geiger, E., Blinder, J., Benson, D. W., Goldmuntz, E. (2003). NKX2.5 mutations in patients with congenital heart disease. Journal of the American College of Cardiology, 42(9), 1650–1655. DOI 10.1016/j.jacc.2003.05.004. [Google Scholar] [CrossRef]
30. Page, D. J., Miossec, M. J., Williams, S. G., Monaghan, R. M., Fotiou, E. et al. (2019). Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of fallot. Circulation Research, 124(4), 553–563. DOI 10.1161/CIRCRESAHA.118.313250. [Google Scholar] [CrossRef]
31. Wu, Q., Wang, H., He, F., Zheng, J., Zhang, H. et al. (2022). Depletion of microRNA-92a enhances the role of sevoflurane treatment in reducing myocardial ischemia-reperfusion injury by upregulating KLF4. Cardiovascular Drugs and Therapy, 22(10), 4886. DOI 10.1007/s10557-021-07303-x. [Google Scholar] [CrossRef]
32. Zhang, Y., Da, Q., Cao, S., Yan, K., Shi, Z. et al. (2021). HINT1 (Histidine Triad Nucleotide-Binding Protein 1) attenuates cardiac hypertrophy via suppressing HOXA5 (Homeobox A5) expression. Circulation, 144(8), 638–654. DOI 10.1161/CIRCULATIONAHA.120.051094. [Google Scholar] [CrossRef]
33. Forini, F., Nicolini, G., Kusmic, C., D’Aurizio, R., Mercatanti, A. et al. (2020). T3 critically affects the Mhrt/Brg1 axis to regulate the cardiac MHC switch: Role of an epigenetic cross-talk. Cells, 99(10), 2155. DOI 10.3390/cells9102155. [Google Scholar] [CrossRef]
34. Rutland, C., Warner, L., Thorpe, A., Alibhai, A., Robinson, T. et al. (2009). Knockdown of alpha myosin heavy chain disrupts the cytoskeleton and leads to multiple defects during chick cardiogenesis. Journal of Anatomy, 214(6), 905–915. DOI 10.1111/j.1469-7580.2009.01079.x. [Google Scholar] [CrossRef]
35. Xia, Y., Chen, S., Yang, Y., Wu, Y., Huang, S. et al. (2019). Novel mutation in MYH6 in 2 unrelated Chinese Han families with familial atrial septal defect. Circulation: Genomic and Precision Medicine, 12(11), e002732. DOI 10.1161/CIRCGEN.119.002732. [Google Scholar] [CrossRef]
36. Park, S., Luk, S., Bains, R. S., Whittaker, D. S., Chiem, E. et al. (2021). Targeted genetic reduction of mutant huntingtin lessens cardiac pathology in the BACHD mouse model of huntington’s disease. Frontiers in Cardiovascular Medicine, 8, 810810. DOI 10.3389/fcvm.2021.810810. [Google Scholar] [CrossRef]
37. Zheng, S. Q., Chen, H. X., Liu, X. C., Yang, Q., He, G. W. (2021). Identification of variants of ISL1 gene promoter and cellular functions in isolated ventricular septal defects. American Journal of Physiology-Cell Physiology, 321(3), C443–C452. DOI 10.1152/ajpcell.00167.2021. [Google Scholar] [CrossRef]
38. Andersson, R., Sandelin, A. (2020). Determinants of enhancer and promoter activities of regulatory elements. Nature Reviews Genetics, 21(2), 71–87. DOI 10.1038/s41576-019-0173-8. [Google Scholar] [CrossRef]
39. Pashmforoush, M., Lu, J. T., Chen, H., Amand, T. S., Kondo, R. et al. (2004). Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell, 117(3), 373–386. DOI 10.1016/S0092-8674(04)00405-2. [Google Scholar] [CrossRef]
40. Schott, J. J., Benson, D. W., Basson, C. T., Pease, W., Silberbach, G. M. et al. (1998). Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science, 281(5373), 108–111. DOI 10.1126/science.281.5373.108. [Google Scholar] [CrossRef]
41. Zakariyah, A. F., Rajgara, R. F., Veinot, J. P., Skerjanc, I. S., Burgon, P. G. (2017). Congenital heart defect causing mutation in Nkx2.5 displays in vivo functional deficit. Journal of Molecular and Cellular Cardiology, 105, 89–98. DOI 10.1016/j.yjmcc.2017.03.003. [Google Scholar] [CrossRef]
42. Dixon, J. E., Dick, E., Rajamohan, D., Shakesheff, K. M., Denning, C. (2011). Directed differentiation of human embryonic stem cells to interrogate the cardiac gene regulatory network. Molecular Therapy, 19(9), 1695–1703. DOI 10.1038/mt.2011.125. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools