
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
Comparative Transcriptome Analysis of Seed Germination of a Cotton Variety with High Tolerance to Low Temperature
1 College of Agriculture, Henan Institute of Science and Technology, Xinxiang, 453003, China
2 College of Life Sciences, Henan Institute of Science and Technology, Xinxiang, 453003, China
* Corresponding Author: Genhai Hu. Email:
Phyton-International Journal of Experimental Botany 2023, 92(9), 2535-2554. https://doi.org/10.32604/phyton.2023.030163
Received 24 February 2023; Accepted 15 May 2023; Issue published 28 July 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Gossypium hirsutum L. is an important cash crop native to the subtropics and is widely cultivated around the world. Low temperature is an important stress that seriously affects seed germination and emergence during planting. In this study, transcriptomic profiles of low-temperature- and normal-temperature-germinated seeds of Xinluzao 25, a variety with low-temperature tolerance and high germination rates, were analyzed and compared. The following results were obtained. (1) A total of 81.06 Gb of clean data were obtained after transcriptome sequencing and assembly, and 76,931 non-redundant Unigene sequences were obtained after data consolidation and concatenation; of these, 69,883 Unigene sequences were annotated. In addition, 55,463 Unigene transcript sequences (72.2%) were annotated for Gene Ontology (GO) classification, and 26,629 genes were involved in 50 metabolic pathways identified by Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. (2) Three main pathways related to low-temperature tolerance of seed germination were identified: starch and sucrose metabolism, phenylpropanoid biosynthesis, and cysteine and methionine metabolism. Their main molecular functions involve the regulation of abscisic acid and activities of enzymes such as amylase, peroxidase, and oxidoreductase. During germination at low temperature, more genes were down-regulated than up-regulated genes at the protrusion stage (2 mm), and more genes were up-regulated than down-regulated at the germination stage (30 mm) after protrusion. (3) The enzyme activities at the two stages showed that amylase, peroxidase, catalase, and glutathione reductase had higher activities when the seeds germinated at 15°C. In this study, high expression of amylase, peroxidase, catalase, and glutathione reductase genes may be the main cause of increased tolerance to low temperature.Keywords
Supplementary Material
Supplementary Material FileUpland cotton is an important fiber crop native to subtropical regions and prefers warm temperatures [1,2]. China is an important cotton producer. As the population increases, more land is needed to grow food crops. Therefore, China’s planting structure is continually adjusting. With the adjustment of China’s agricultural planting structure, cotton growing areas have gradually moved northward, and southern Inner Mongolia and Xinjiang have become the main cotton growing areas in China [3]. Barpete reported that 30°C was optimal for early cotton seed germination, and seeds germinated slowly at 20°C [4]. When the ambient temperature is below 15°C, seeds cannot germinate normally [5]. In Xinjiang, spring weather is often cold, requiring many cotton fields to be replanted. To ensure its growth, cotton is often sown with mulch in early spring when the temperature is below 12°C [6], and early sowing of cotton is generally from April 5 to 18 each year, when the average daily temperature is 17.5°C and mulch is no longer required [7]. The use of mulch technology for long periods leads to serious pollution in cotton fields because of its residual film [8]. Only normal seed germination and seedling development can ensure a good harvest [9]. Therefore, it is very important to understand the mechanism of low-temperature tolerance in cotton seed germination.
When plants were moved from the normal to the low-temperature environment, a series of physiological and biochemical reactions were initiated, in which a fine regulatory network provided low-temperature protection [10]. Glycine, betaine, proline, soluble sugars, and polyols increase significantly after plants were transferred to low-temperature environments [11,12]. From the perspective of a metabolic pathway, low-temperature environments induce new gene expression in plants, which encode stress-response proteins, including various transcription factors, protein kinases, and other signaling molecules [13]. Many transcription factors are involved in low-temperature stress responses in plants, and the ICE–CBF–COR signaling pathway is the most well understood mechanism [14]. In addition to the CBF cold reaction pathway, there are many other low-temperature regulatory pathways, and many genes are down-regulated during cold acclimation. It is possible that gene suppression plays an indispensable role in cold acclimation [15].
Seed germination is the beginning of a new plant life cycle; according to water absorption rates, seed germination is divided into three phases [16]. Imbibition is the first stage of seed germination and is mainly a physical phenomenon caused by water absorption by large numbers of hydrophilic colloids contained in dry seeds. Protrusion is the second stage of seed germination, and the seeds stop absorbing water. During this period, activation of biological macromolecules and organelles leads to rapidly increased enzyme activity in the seeds, respiration is enhanced, nutrient metabolism is vigorous, and the radicle begins to grow through the seed coat. Germination is the third stage and is characterized by rapid elongation of the radicle germ. Low temperatures increase the duration, or even stop, protrusion and germination [17,18]. Therefore, it is important to understand the regulatory mechanisms of seed germination.
The transcriptome is the collection of all transcripts produced by a certain species or specific cell type, reflecting differences in gene expression under different physiological states and environmental conditions [19]. Transcriptome analysis of seed germination tolerance to low temperature has been studied in some plants, such as maize [20], Ricinus communis [21], Platycodon grandiflorum [22], and upland cotton [23]. Shen et al. conducted transcriptome analysis of seed germination at different times under low-temperature treatment for two cotton varieties with different cold tolerances. The results showed that the main differences between the two varieties appeared in glycolysis/gluconeogenesis, the tricarboxylic acid cycle, and the glyoxylate cycle. Additional related hormone content determination and gene expression analysis indicated that Indoleacetic acid (IAA), cytokinin (CTK), and gibberellin (GA3) may have promoted germination in the cold-tolerant variety [23]. The two varieties with different cold-tolerance sensitivities had different genetic backgrounds. Therefore, we compared the transcriptome of a single cold-tolerant variety at the same germination stage at low temperature and room temperature.
Khetran et al. reported that the lowest seed germination rate was observed at 15°C [18], but the variety Xinluzao 25, which is low-temperature resistant with a high germination rate at 15°C, was found [24]. In our study, germination experiments with Xinluzao 25 at 15°C and 25°C were performed to compare the transcriptomes during protrusion and germination. This study was based on transcriptome sequence analysis. The differences in gene transcription and expression of low-temperature-tolerant cotton seeds during low-temperature and normal-temperature germination were compared; the biological functions were also analyzed, and the molecular mechanism of low-temperature-tolerant cotton seed germination was discussed, which will provide a foundation for improving the low-temperature-tolerant cotton seed germination through breeding. The activities of amylase, peroxidase (POD), catalase (CAT), and glutathione reductase (GR) related to seed germination at two stages were measured to detect their corresponding relationships with the transcriptome sequencing results.
2.1 Plant Materials and RNA Extraction
Xinluzao 25, a variety with a high germination rate at low temperatures, was selected as the experimental material. Full seeds were selected and delinted with sulfuric acid. Six duplicate seeds were selected, each containing 100 seeds. The seeds were soaked in 0.1% mercury chloride for 15 min for disinfection and washed with distilled water five times. The seeds were uniformly discharged into germinating boxes padded with double layers of filter paper, and 50 mL purified water was added to each germinating box. After 22 h, the seeds were saturated with water, and the water was poured out. Then 3 to 5 mL of purified water were added to keep the filter paper moist. Three germination boxes were placed in a 15°C incubator for the germination test. The other three boxes were placed in a constant-temperature incubator at 25°C. Samples were taken from the seed radicles at when they were 2 and 30 mm long (Fig. 1), and each was repeated three times. The seed coat was peeled off with sterilized tweezers while wearing latex gloves, and the samples were packaged and labeled (2-mm samples were labeled L251 for radicle germination, and the 30-mm samples were labeled L252 for incubator germination at 15°C; incubators at 25°C contained 2-mm samples labeled H251 radicle and 30-mm samples labeled H252 radicle). The samples were stored in liquid nitrogen for later use. In this research, the 15°C treatment is described as the low-temperature treatment, and the 25°C treatment is described as the normal-temperature treatment.
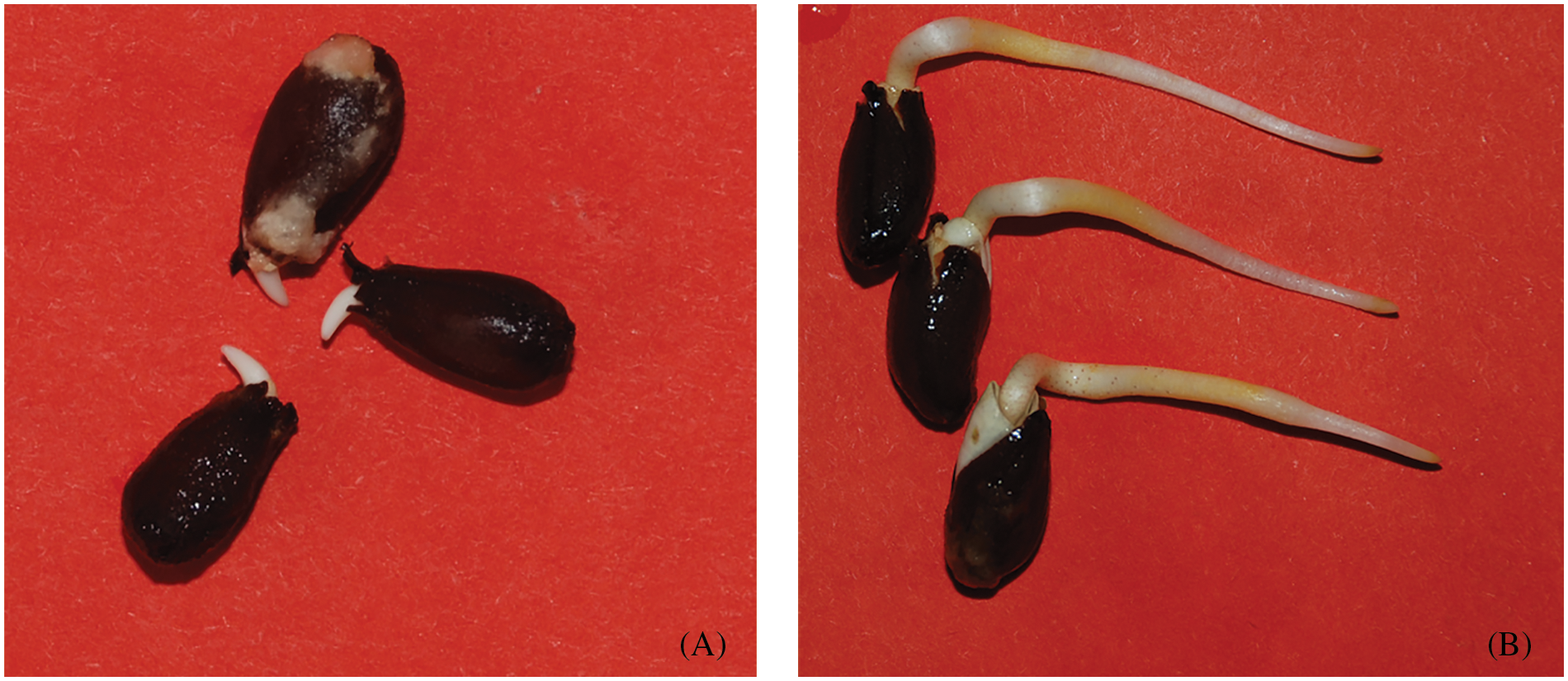
Figure 1: Sampling phase pictures. (A) Seeds protrusion radicle 2 mm; sampling time, 4 days after imbibition of seeds at 15°C and 1 day at 25°C; (B) seeds germinate radicle 30 mm, sampling time, 14 days after imbibition of seeds at 15°C and 3 days at 25°C
Total RNA from germinated cotton seeds was extracted using a Qiagen 70022 oligotex mRNA mini kit (QIAGEN, Strasse 1 40724 Hilden, Germany). RNA sample purity, concentration, and integrity was detected using Nanodrop (IMPLEN, USA), Qubit2.0 (Life Technologies, USA), and Aglient2100 (Agilent Technologies, USA) machines.
2.2 cDNA Preparation and Illumina Sequencing
Three replicates were collected for each period, and mRNA samples from 12 materials were obtained. Using qualified mRNAs as templates, cDNA libraries were obtained using tools and techniques including random hexamers, AMPure XP beads, and PCR enrichment. Qubit2.0, Agilent 2100, a quantitative reverse transcription and polymerase chain reaction (qRT-PCR) thermocycler, and other instruments and methods were used to ensure the quality of the constructed library. After qualification, the Illumina Hiseq was used for high-throughput sequencing.
2.3 Sequence Data Filtering and De Novo Assembly
Raw reads were processed to obtain high-quality clean reads. The final Unigenes were obtained using Trinity assembly software, the de Bruijn mapping method, and sequencing read information [25].
Note on Unigenes
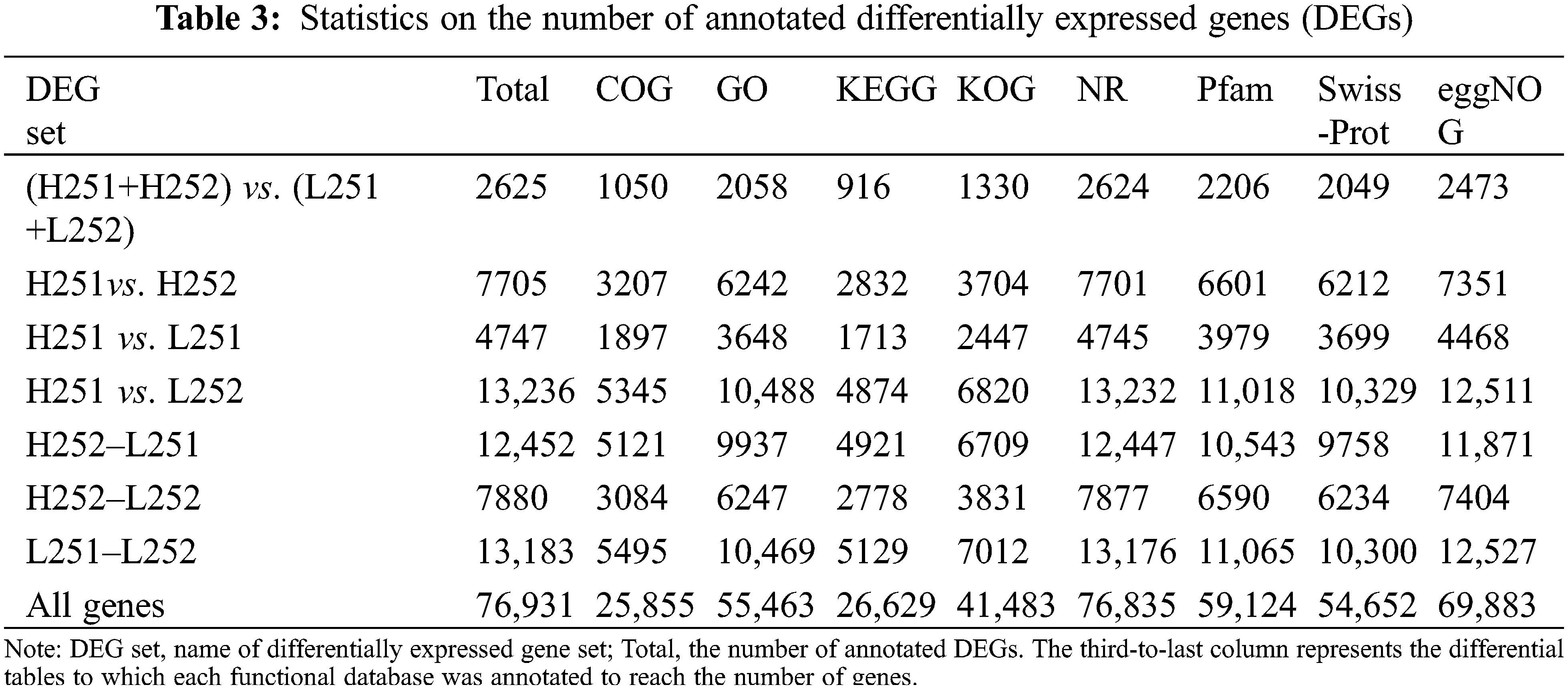
Sequences of differentially expressed genes (DEGs) were sequentially aligned to Clusters of Orthologous Groups (COG) by BLAST. Sequences were also aligned with the Gene Ontology (GO), KEGG, eu-Karyotic Orthologous Groups (KOG), Protein Family (Pfam), Swiss-Prot (Manually Annotated and Reviewed Protein Sequence Database, Swiss-Prot), eggNOG database (Evolutionary Genealogy of Genes, Non-supervised Orthologous Groups, eggNOG) and Non-redundant (NR) databases, and related information was obtained.
Quantitative analysis of expression difference
The Fragments Per Kilobase of transcript per Million fragments mapped (FPKM) value was used to represent the expression abundance of Unigenes. The selection criteria for DEGs were the following: False Discovery Rate (FDR) < 0.05 and log2|FC| ≥ 2, including the fold change (FC), which is an expression of the ratio of volumes between the two samples. GO functional enrichment analysis, KEGG annotation, and pathway enrichment analysis were performed for the DEGs.
2.4 Verification of Enzyme Activity at Two Stages of Seed Germination
To verify the results of the transcriptome comparisons for seed germination at low temperature and normal temperature, the activities of enzymes important during seed germination at low temperatures were measured. For the two temperatures, three seeds with 2-mm radicles and three seeds with 30-mm radicles were collected. The seed coats were peeled off and weighed, and this was repeated three times. Alpha-amylase activity was determined by spectrophotometry. Peroxidase was determined by the guaiacol method, and the activity of peroxidase was determined by potassium permanganate titration [26]. Glutathione reductase activity was determined by spectrophotometry [27]. Data analysis was assisted by microsoft Office Excel software (Microsoft (China) Co., Ltd., 5 Danleng Street, Haidian District, Beijing).
2.5 Analysis of the Four Enzymes by qRT-PCR
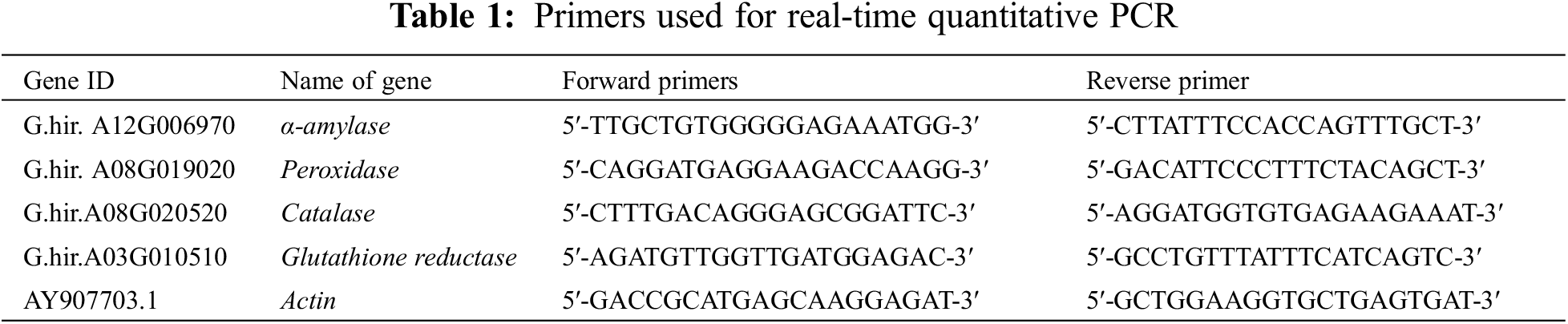
The gene Actin (GenBank accession number: AY907703.1) was used as the reference gene. Four DEGs identified in the transcriptome were selected, including one each for amylase, peroxidase, catalase and glutathione reductase. Quantitative real-time PCR (qRT-PCR) analysis was performed. The qRT-PCR primer sequences are shown in the Table 1. Three biological replicates were used for each sample, qRT-PCR was performed using the TB Green Premix Ex Taq II kit (Takara Biomedical Technology (Beijing) Co., Ltd., 22 Science Park Road, Changping District, Beijing, China) and the 2−ΔΔCt method was used to calculate the relative gene expression.
3.1 Differential Gene Expression Analysis
The Pearson correlation coefficient was used to assess the correlation between the sample evaluation indexes [28]. The closer r2 is to 1, the stronger the correlation is between the two samples. According to correlation analysis, r2 values between L251-1, L251-2, and L251-3 and H251-1, H251-2, and H251-3 were all >0.900, indicating strong correlation between L251-1, L251-2, and L251-3 and H251-1, H251-2, and H251-3. Proof that the gene expression level was stable at the time point of examination can be used for subsequent analysis (Fig. 2A).
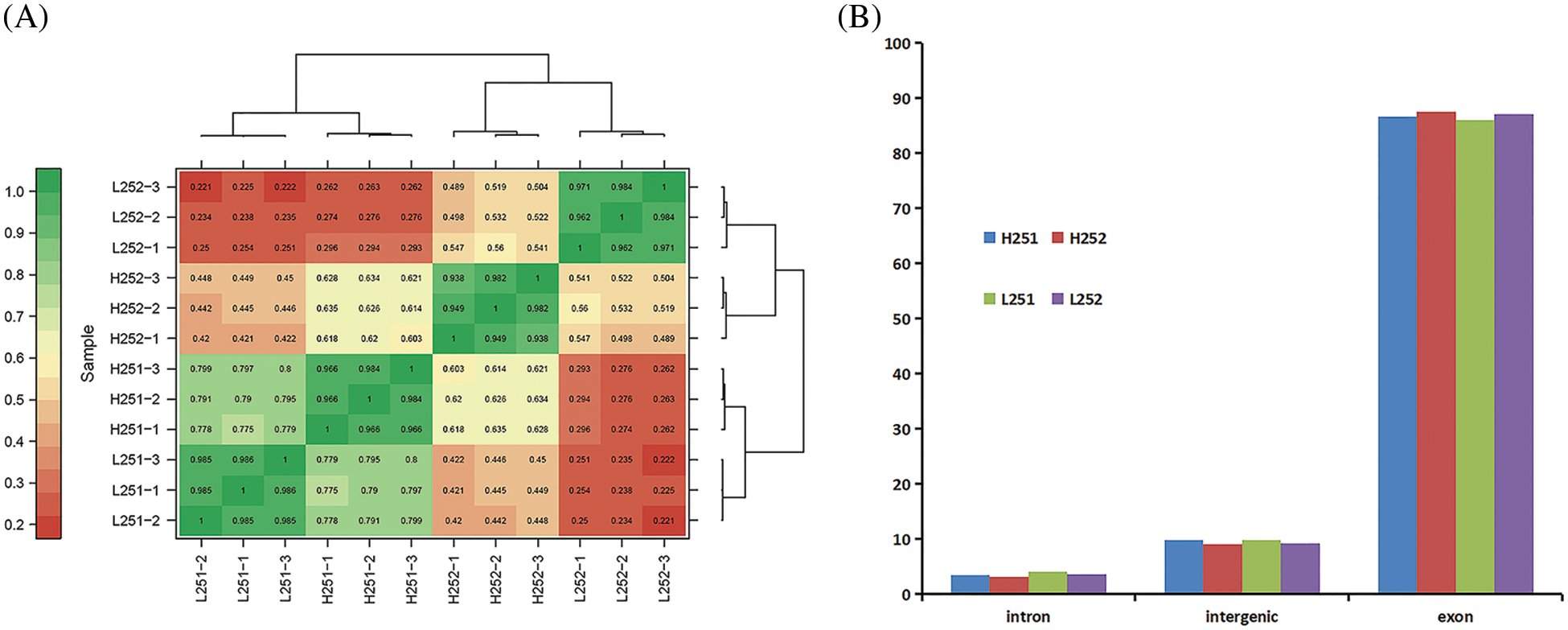
Figure 2: Correlation analysis and distribution of mapped reads of gene expression in each sample group. (A) Correlation analysis of gene expression in each sample group, the larger the value in the figure, the higher the correlation between samples; L251-1–L252-3 and H251-1–H252-3: sample numbers. (B) Distribution of mapped reads in different regions of the genome, the figure divides the genome into exonic, intergenic, and intronic regions
Transcriptome analysis of 12 samples was completed. A total of 81.06 Gb of clean data were obtained. The clean data of all samples reached 6.02 Gb, and the percentage of Q30 bases was 93.78% or above (Table S1). Sequence alignment between the clean reads of each sample and the designated reference genome was performed, and the alignment efficiency ranged from 91.49% to 94.77%. Based on the results of the comparisons, predictive variable-splicing analysis, gene structure optimization analysis, and new gene discovery were conducted. 8516 new genes were discovered, of which 7,407 were functional annotations. Based on the comparison results, gene expression was analyzed. DEGs were identified according to their expression levels in different samples, and functional annotations and enrichment analyses were performed.
3.2 Type Analysis of Expressed Genes
The gene expression types of each sample were classified as intronic, intergenic, or exonic. The numerical proportion of gene expression in samples at different stages was counted (Fig. 2B). The results showed that intron expression, intergenic expression, and exonic expression accounted for 3.18%–4.05%, 9.10%–9.84%, and 86.07%–87.63% of gene expressions, respectively. According to Fig. 2B, most genes expressed in cotton seed germination were exons and the fewest were expressed were introns. Both the intronic and intergenic proportions decreased with radicle elongation, but the exonic proportions increased with increasing radicle length at both low and normal temperatures. Compared with the expressions at normal temperature, the expression of introns was slightly higher, and that of exons was slightly lower at low temperature.
3.3 Statistics about the Numbers of Differentially Expressed Genes
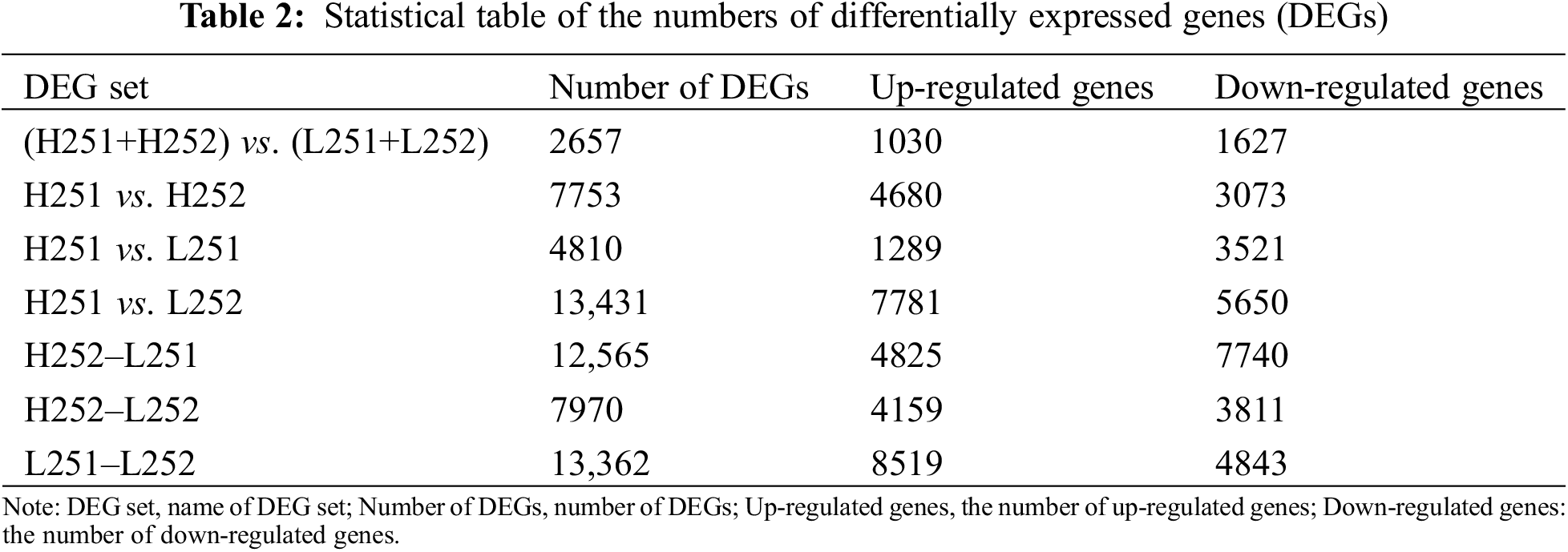
The number of DEGs in Xinluzao 25 was 2657: 1030 were up-regulated, and 1627 were down-regulated under both low temperature and normal temperature. Under the two conditions, the number of DEGs at the protrusion stage was 4810, with 1289 up-regulated and 3521 down-regulated genes. Under the low-temperature condition, the number of DEGs that continued to be expressed during radicle elongation after protrusion was 13,362, with 8519 up-regulated and 4843 down-regulated genes. Under normal-temperature conditions, 7753 differential genes were expressed after protrusion, of which 4680 were up-regulated, and 3073 were down-regulated. Under the conditions of low temperature and normal temperature, 7970 differential genes were expressed after protrusion: 4159 were up-regulated, and 3811 were down-regulated (Table 2). According to metabolic pathway analysis, when H251 and L251 samples were compared, the main up-regulated genes were enriched in plant hormone signal transduction, purine metabolism, spliceosomes, starch and sucrose metabolism, and the down-regulated genes were mainly concentrated in carbon metabolism, phenylpropanoid biosynthesis, starch and sucrose metabolism, and plant hormone signal transduction. In the comparison between H252 and L252 samples, the main up-regulated genes were chiefly concentrated in plant hormone signal transduction, carbon metabolism, photosynthesis, and biosynthesis of amino acids; the down-regulated genes were enriched in phenylpropanoid biosynthesis, plant hormone signal transduction, pentose and glucuronate interconversions, and carbon metabolism. In the comparison between L252 and L251, the main up-regulated genes were enriched in carbon metabolism, plant hormone signal transduction, biosynthesis of amino acids, and phenylpropanoid biosynthesis, and the down-regulated genes were enriched in ribosomes, plant hormone signal transduction, ribosome biogenesis in eukaryotes, purine metabolism, and carbon metabolism.
3.4 Functional Annotation and Enrichment Analysis of Differentially Expressed Genes
A total of 76,931 Unigenes was obtained. The entirety of the expressed gene sequences was compared with the COG, GO, KEGG, KOGs, Pfam, Swiss-Prot, eggnog, and NR databases. The lowest annotation rate of the Unigene was in the COG database at 33.6%, and the highest annotation rate was in eggNOG at 90.8% (Table 3).
3.5 Gene Ontology (GO) Enrichment Analysis
According to the differential genes screened, determining their distribution in the GO categories will help clarify sample differences in gene function. In GO enrichment analysis, there are three main categories: biological processes (BP), cellular components (CC), and molecular functions (MFs). The statistical results of the GO classification of DEGs are shown in Fig. 3.
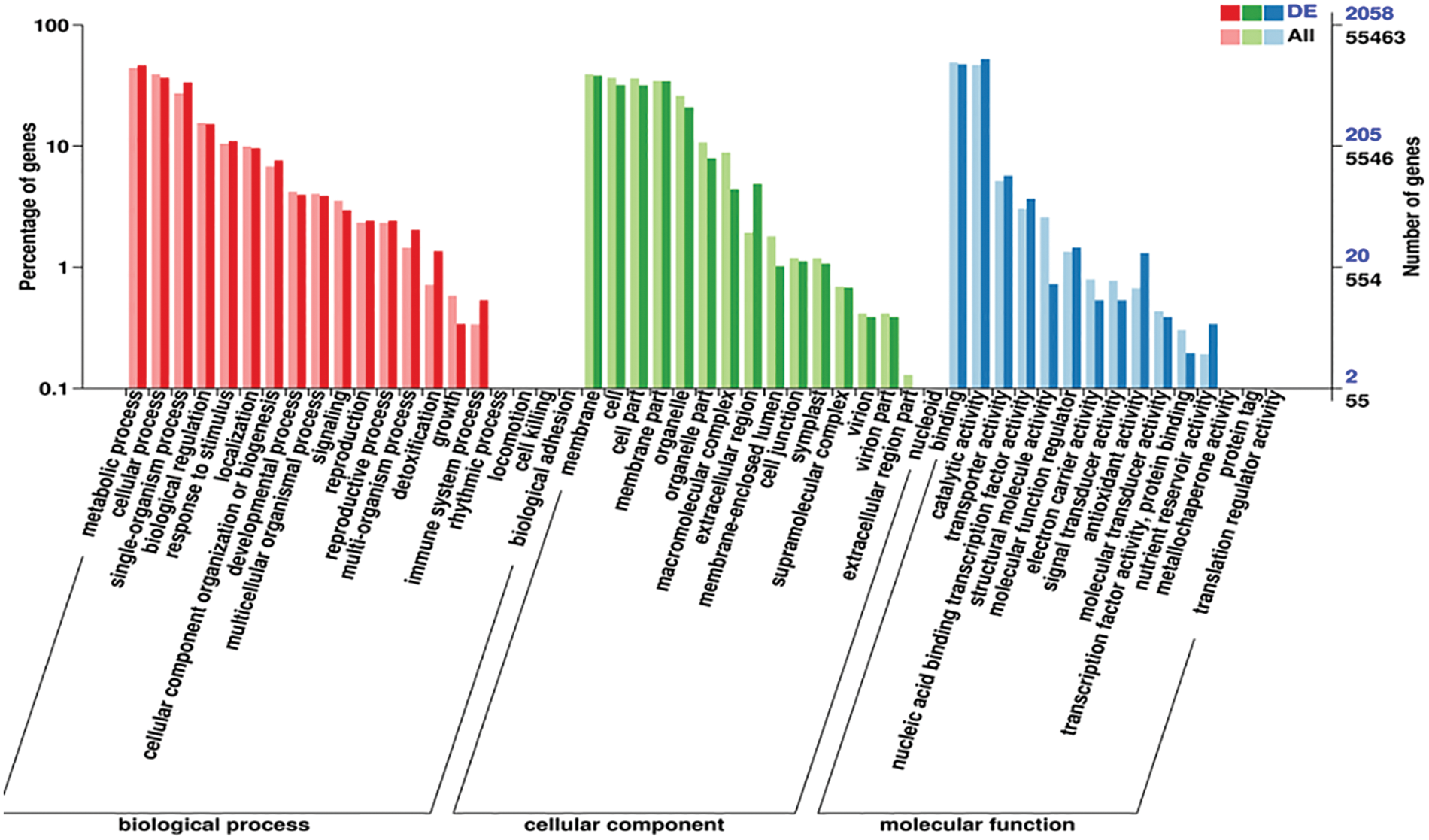
Figure 3: Statistical map of GO annotation classification of differentially expressed genes
Note: The horizontal coordinate represents the GO classification, the left side of the vertical coordinate is the percentage of the number of genes, and the right side is the number of genes
The results of differential gene cluster analysis showed that differential genes are mainly enriched in metabolic processes, cellular processes, and single-organism processes in BP; differential genes are mainly enriched in membranes, cells, cell parts, and membrane parts in CC; and differential genes are mainly concentrated in binding and catalytic activity in MF.
To understand the differences in up-regulated and down-regulated genes between low temperature and normal temperature germinated seeds, the up-regulated and down-regulated functional genes of H251 vs. L251, H252 vs. L252, and L251 vs. L252 were compared and analyzed (Fig. 4). In the H251 vs. L251 comparison, the up-regulated genes were mainly enriched in such processes as RNA binding, helicase activity, ATPase activity, starch synthase activity, and abscisic acid 8′-hydroxylase, and the down-regulated genes were mainly enriched processes involving metal-ion binding, heme binding, iron-ion binding, oxidoreductase activity, and monooxygenase activity. In the H252 vs. L252 comparison, the up-regulated genes were mainly enriched in processes involving metal-ion binding, transcription factor activity, oxidoreductase activity, monooxygenase activity, and chlorophyll binding; the down-regulated genes were mainly enriched in processes involving metal-ion binding, heme binding, iron-ion binding, oxidoreductase activity, and monooxygenase activity. In the L251 vs. L252 comparison, the up-regulated genes were mainly enriched in processes involving heme binding, iron-ion binding, oxidoreductase activity, monooxygenase activity, and peroxidase activity; the down-regulated genes were mainly enriched in processes involving the structural constituents of ribosomes, transcription factor activity, sequence-specific DNA binding, and peroxidase activity.
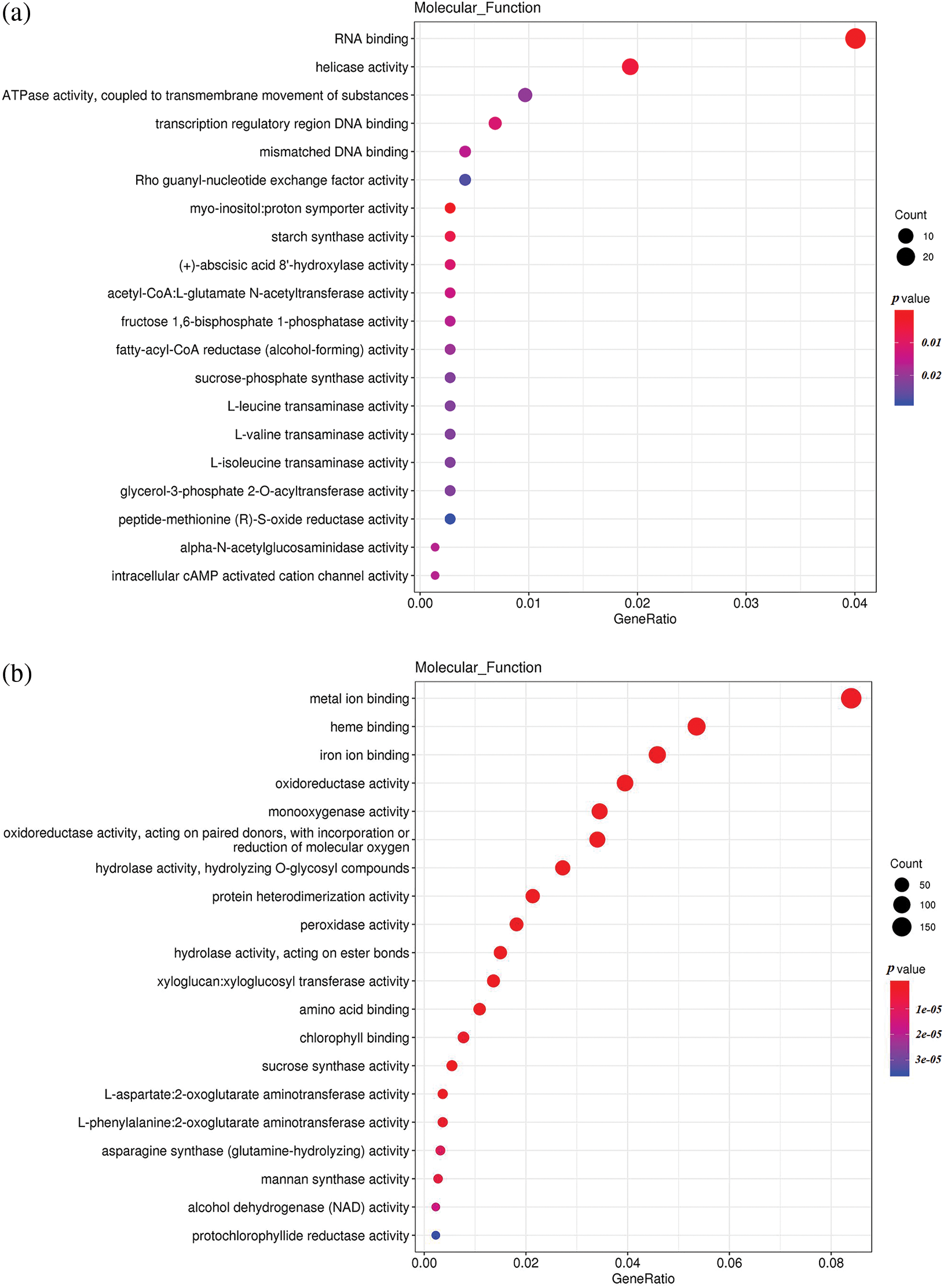
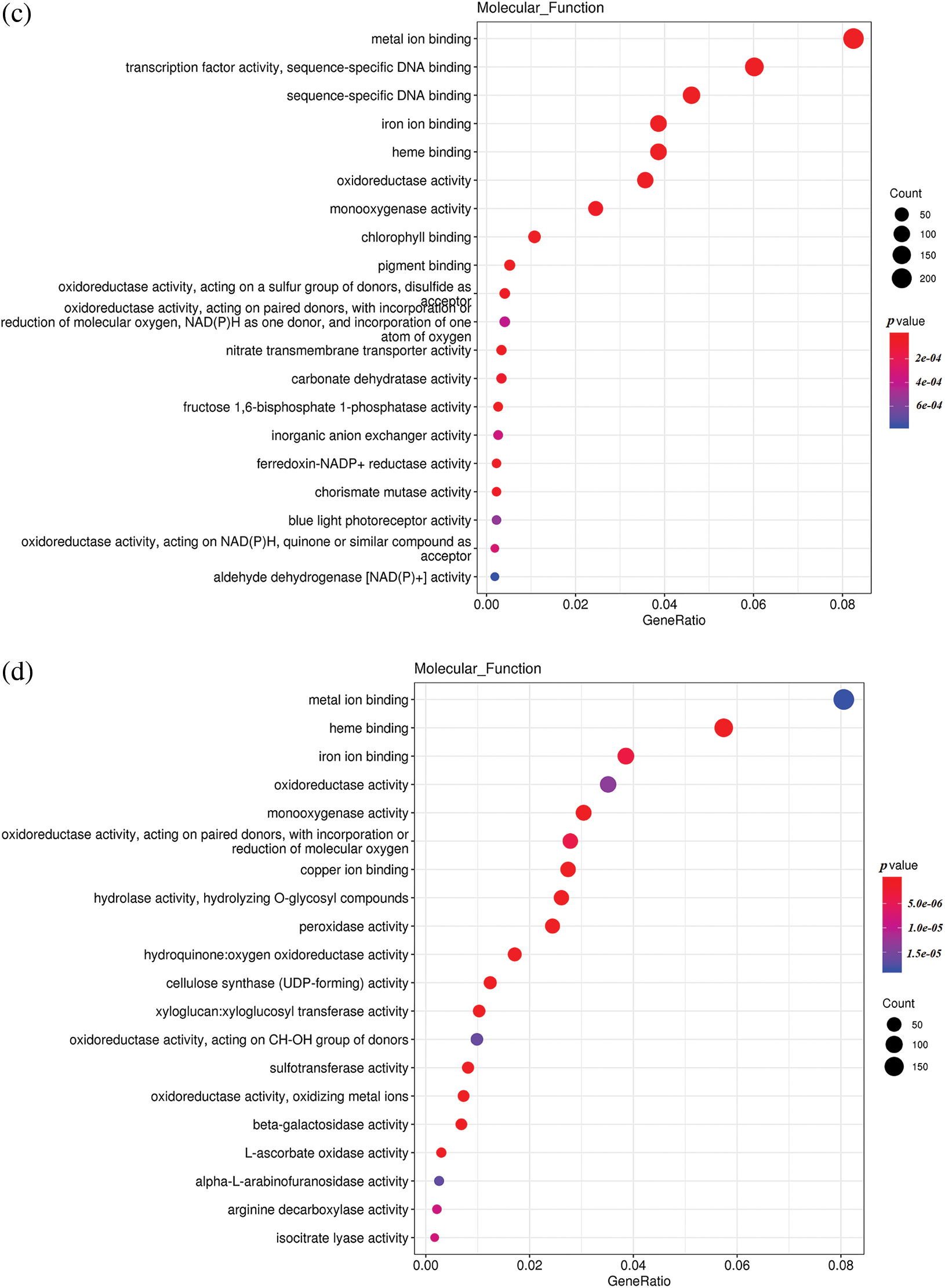
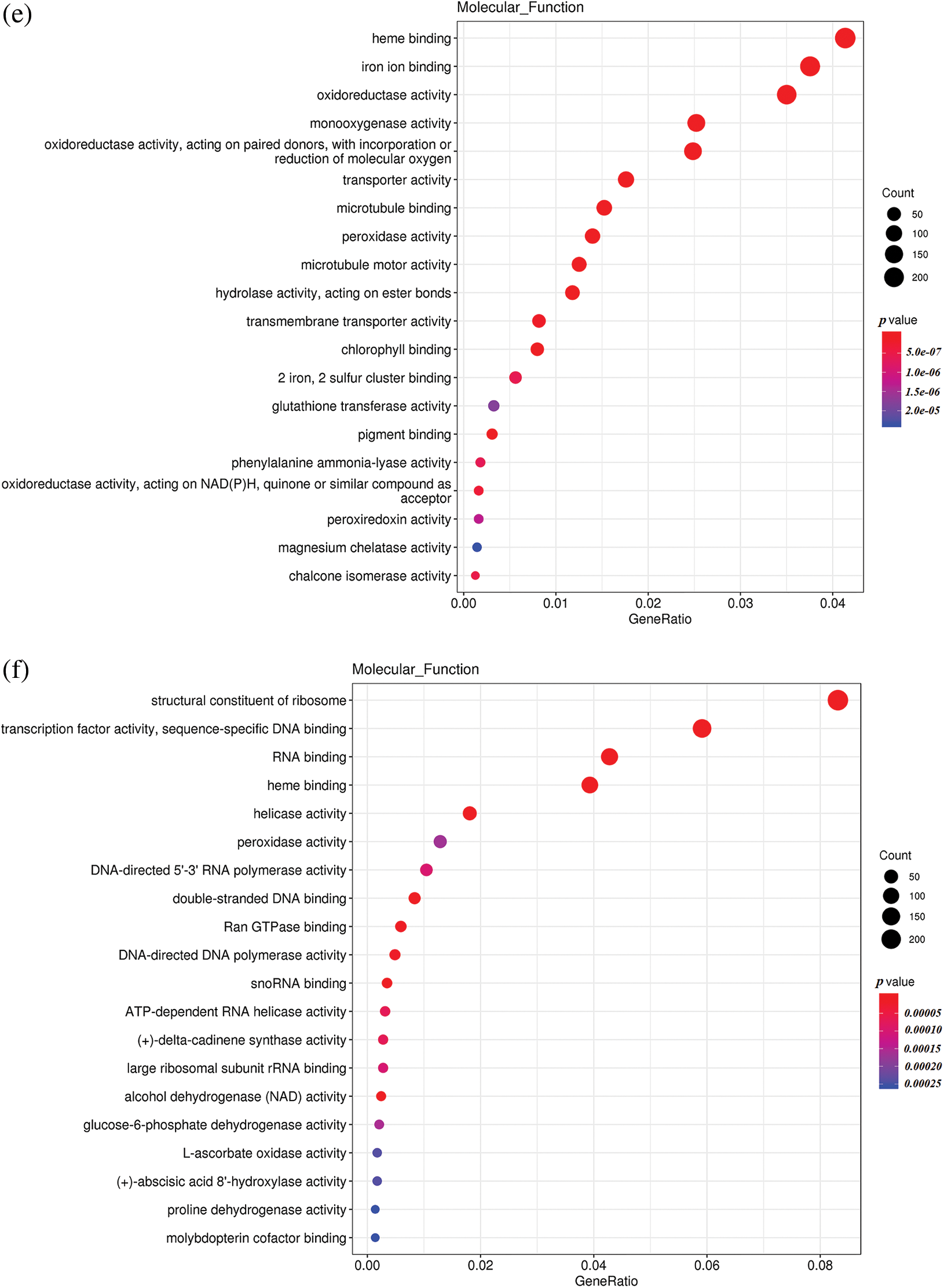
Figure 4: Scatter plot of GO enrichment analysis of the molecular functions of differentially expressed genes
Note: The horizontal coordinate is the GeneRatio, indicating the proportion of genes in this entry compared with all differentially expressed genes (DEGs), and the ordinate coordinate represents each molecular function. The size of the dots represents the number of differentially expressed genes annotated in the function, and the color of the dots represents the P value of the hypergeometric test. (a) H251 vs. L251 enrichment of the up-regulated genes; (b) H251 vs. L251 enrichment of the down-regulated genes; (c) H252 vs. L252 enrichment of the up-regulated genes; (d) H252 vs. L252 enrichment of the down-regulated genes; (e) L251 vs. L252 enrichment of the up-regulated genes; (f) L251 vs. L252 enrichment in down-regulated genes
3.6 Analysis of KEGG Metabolic Pathways of Differentially Expressed Genes
Genes exercise their biological functions in coordination with each other. The significant enrichments in KEGG pathways could determine the major biochemical metabolic pathways and signal transduction pathways in which the specific genes are involved [29]. KEGG pathway analysis of the DEGs of Xinluzao 25 indicated they were mainly enriched in starch and sucrose metabolism, phenylpropanoid biosynthesis, and cysteine and methionine metabolism during the germination process at low temperature and normal temperature (Fig. 5).
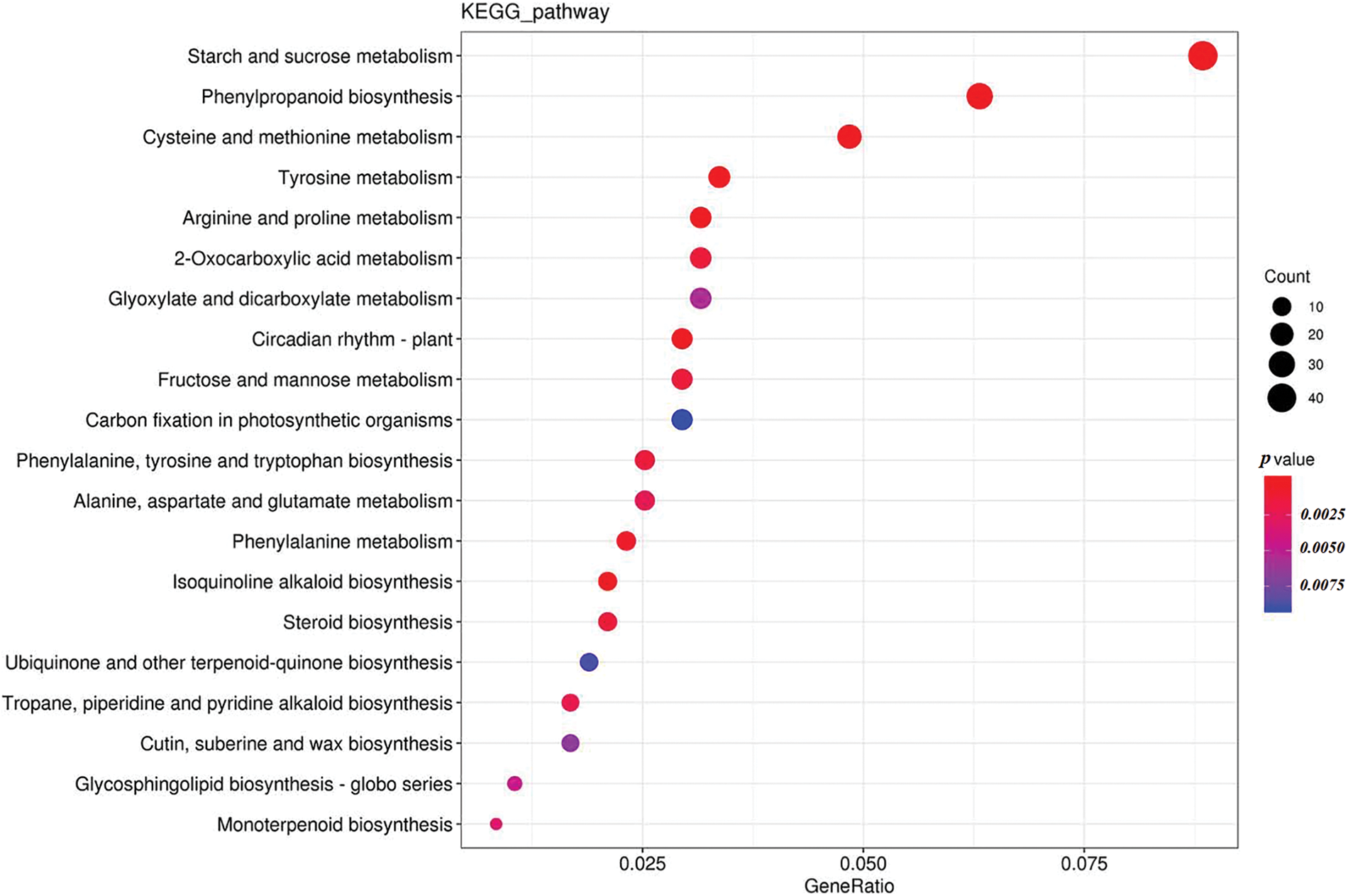
Figure 5: Scatter plot of KEGG enrichment analysis of differentially expressed genes during seed germination of Xinluzao 25 at low temperature and normal temperature
Note: The horizontal coordinate represents the GeneRatio, indicating the proportion of genes in this entry compared with all differentially expressed genes (DEGs), and the ordinate coordinate represents each pathway entry. The size of the dots represents the number of DEGs annotated in the pathway, and the color of the dots represents the p value of the hypergeometric test
The annotation results of the DEGs were classified according to the type of pathway in KEGG. The classification diagram is shown in Fig. 6. KEGG pathways can be divided into five categories: cellular processes, environmental information processing, genetic information processing, metabolism, and organismal systems. Among the above five types of pathways, the pathways that obtained the most hits included starch and sucrose metabolism (42 Unigenes), plant hormone signal transduction (33 Unigenes), biosynthesis of amino acids (32 Unigenes), phenylpropanoid biosynthesis (30 Unigenes), and carbon metabolism (30 Unigenes).
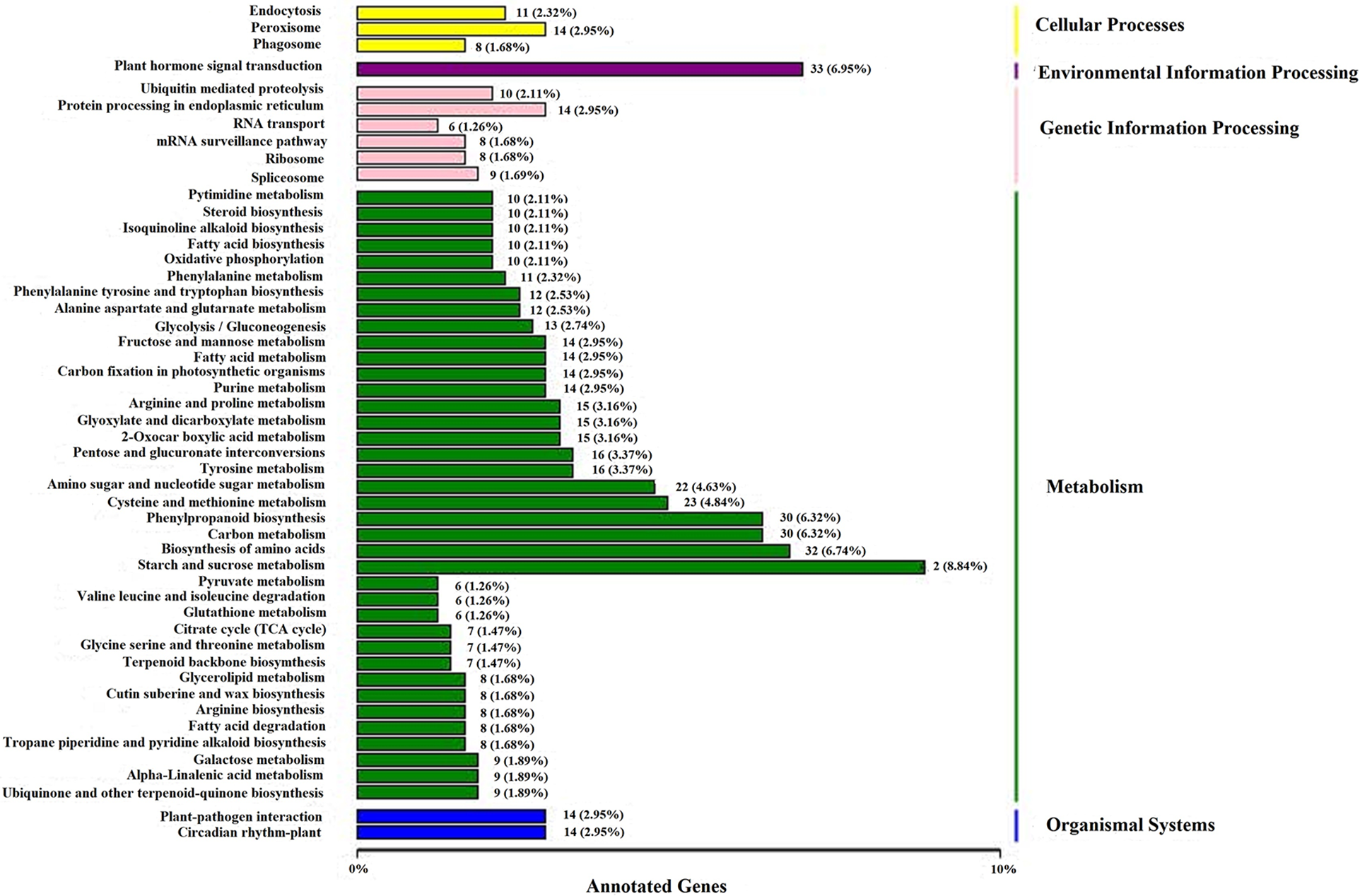
Figure 6: KEGG classification map of differentially expressed genes
Note: The ordinate shows the names of the KEGG metabolic pathways, and the abscissa shows the number of genes annotated to the pathway and their proportion of the total number of annotated genes
3.7 Physiological Response of Seed Germination to Low Temperature
Based on the comparison of the transcriptome data between the two temperatures, the possible relationship between starch and sucrose metabolism, phenylpropanoid biosynthesis, and cysteine and methionine metabolism and low-temperature tolerance was further revealed. Enzyme activity was measured in the radicle during germination (Fig. 7). When the seeds were germinated at low temperature, starch hydrolase, peroxidase, and glutathione oxidase activities were higher whether the radicle was 2 or 30 mm; however, catalase activity was lower at low temperature. Obviously, amylase, peroxidase, and glutathione reductase played more important roles in maintaining metabolic stability and scavenging free radicals in the tissue environment during seed germination at low temperature.
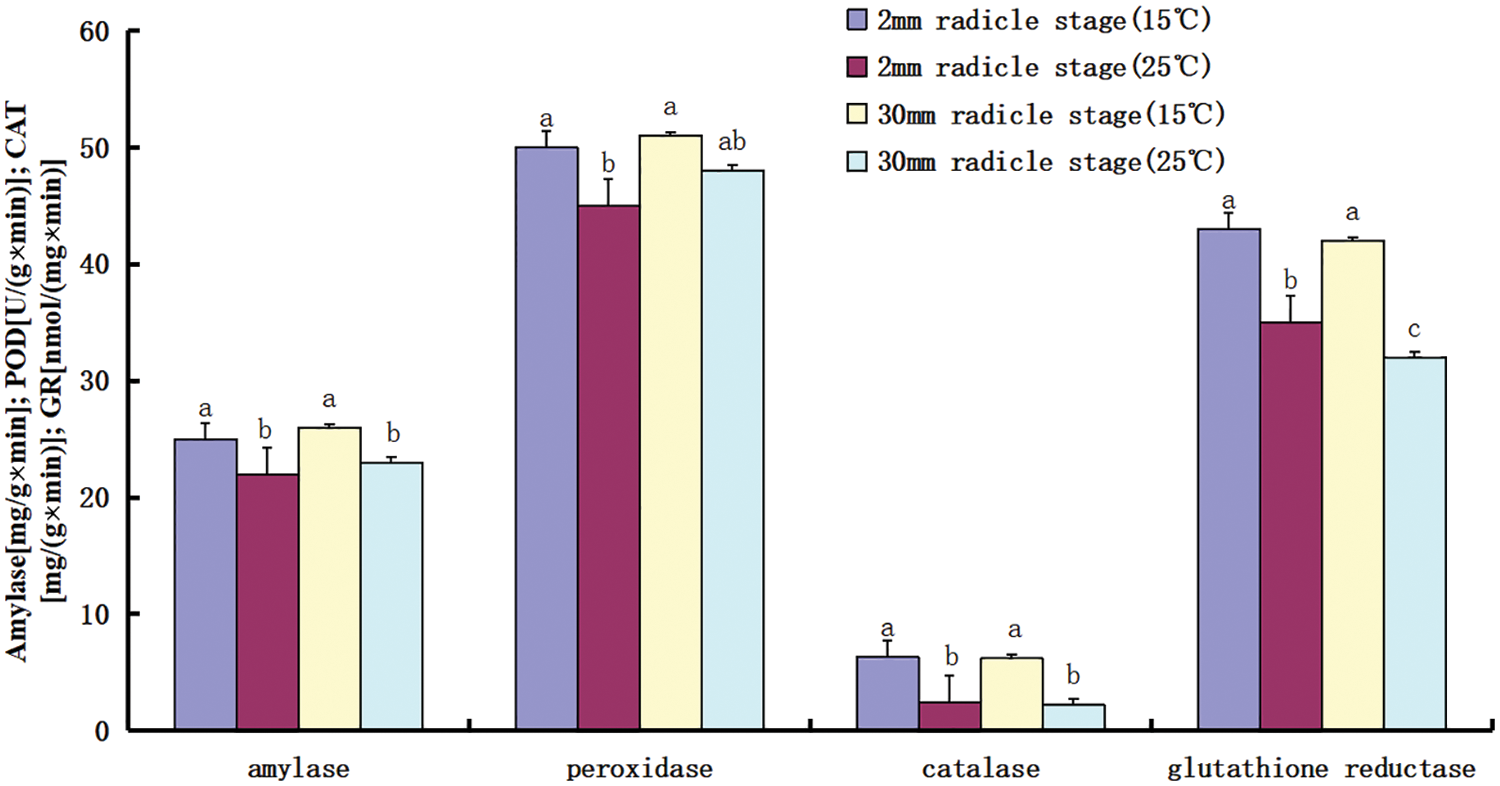
Figure 7: Comparison of enzyme activities during seed germination
Note: The x axis represents the type of enzyme activity measured, and the y axis represents the enzyme activity; activity units are different for each enzyme
3.8 qRT-PCR Validation of Transcriptome Sequencing Data
GO and KEGG analysis based on differentially expressed genes and reaction characteristics of antioxidant protective enzymes during seed germination under low temperature stress. Four differentially expressed genes, including amylase, peroxidase, catalase and glutathione reductase, which were highly correlated with low temperature stress germination, were selected for qRT-PCR verification. The results showed that the expression pattern of qRT-PCR was basically consistent with that of transcriptome sequencing (Fig. 8).
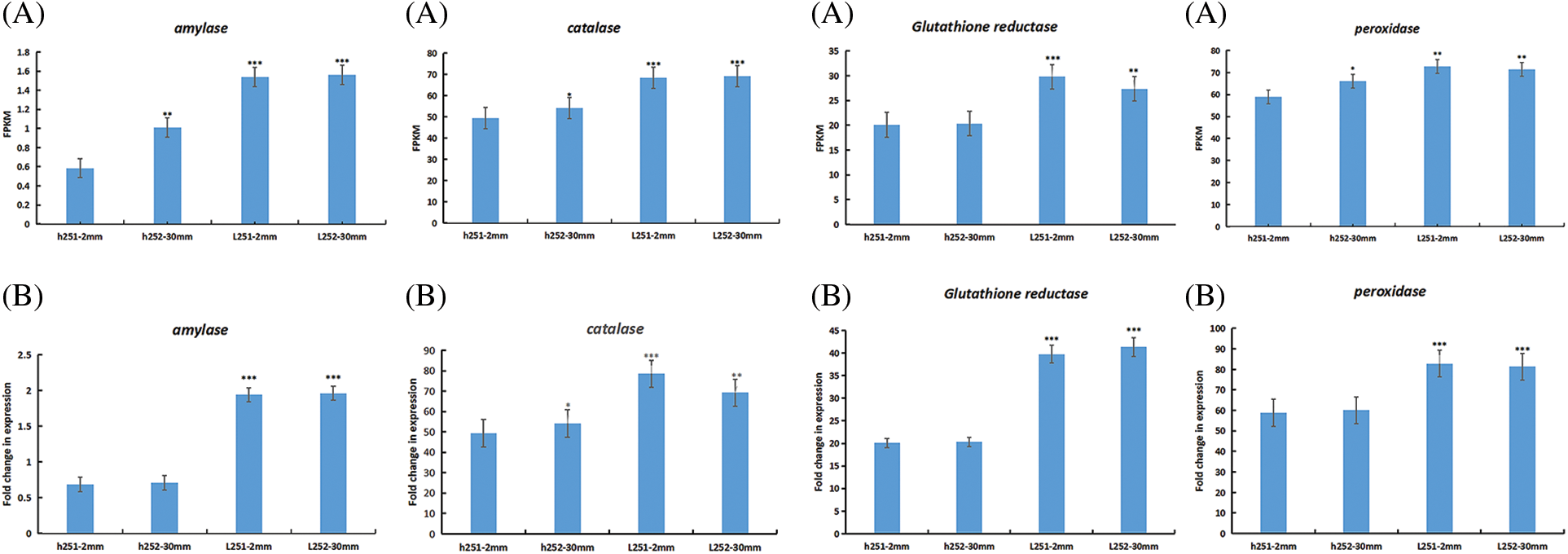
Figure 8: RT-qPCR validation of four differentially expressed genes. (A) Expression by transcriptome sequencing; (B) Relative expression as verified by RT-qPCR. FPKM represents fragment values per kilobase transcription per million mapped reads. *p < 0.05; **p < 0.01; ***p < 0.001
Information about seed germination and seedling vigor is important for cotton development and production. At lower temperatures, the seeds of low-temperature-resistant varieties can develop normally, and cotton field emergence is complete [30,31]. Varieties that are not tolerant to low temperatures often fail to produce normal seedlings, resulting in rotten seeds [17]. The higher germination rate of Xinluzao 25 at low temperature indicated that it had a special ability to respond to low temperatures. In this study, transcriptome analysis was used to compare the changes in Xinluzao 25 response genes at low temperature and normal temperature. The seed germination rate of Xinluzao 25 was still high at low temperature. Under low-temperature stress, Xinluzao 25 could maintain better germination characteristics, which was the result of adaptive gene expression regulation during germination. Transcriptome sequencing showed that 1289 genes were up-regulated, and 3521 genes were down-regulated, at 2-mm protrusion. During radicle elongation after protrusion, 4159 genes were up-regulated, and 3811 genes were down-regulated at 30-mm radicle germination stage. This result is consistent with those of Fower [15]. When the temperature was 15°C, 8519 genes were up-regulated, and 4843 genes were down-regulated, at the 30-mm stage compared with genes at the 2-mm stage. In the same period, there were 4680 up-regulated genes and 3037 down-regulated genes at 25°C. This may indicate that during germination at low temperature, because of the reduced fluidity of cell membranes and decreased enzyme activity in cells, seeds need to sense the signal of low temperature and initiate gene expression to adapt to low temperature, including special genes that allow the needs of seed germination to be met in the low-temperature environment. The results showed that more genes were up-regulated and down-regulated during radicle elongation at low temperature compared with those at normal temperature at the 2-mm protrusion stage and 30-mm germination stage. This indicates that radicle growth requires the expression of more genes to maintain the growth process at low temperature. Lukatkin reported that when cold-sensitive plants encounter low-temperature stress, physiological processes such as water state, mineral nutrition, respiration, and metabolism are disturbed [32]. Our experimental results suggest that gene expression regulation occurred during the germination of Xinluzao 25 at low temperature, and the number of down-regulated genes was greater than the number of up-regulated genes. In addition, the number of up-regulated genes was greater than the number of down-regulated genes when the radicle continued to extend after protrusion. At low temperature, the radicle of germinated seeds requires more up-regulated and down-regulated genes than at normal temperature.
In terms of transcripts, 55,463 of the annotated Unigenes (72.1%) could be classified by GO analysis. There were 2058 DEGs under low- and normal-temperature conditions, indicating that seed germination under low-temperature conditions required the expression of new genes to adapt. There were 3648 DEGs at 15°C at the 2-mm stage compared with genes at 25°C at the 2-mm stage. At this stage, regulatory genes were mainly enriched in starch and sucrose metabolism, plant hormone signal transduction; only up-regulated genes were mainly enriched in purine metabolism and spliceosomes; and only down-regulated genes were mainly enriched in carbon metabolism and phenylpropanoid biosynthesis. There were 6247 DEGs at 15°C at the 30-mm stage compared with genes at 25°C at the 30-mm stage. At this stage, plant hormone signal transduction and carbon metabolism continued to be regulated, and only up-regulated genes were enriched in photosynthesis and biosynthesis of amino acids; only down-regulated genes were enriched in phenylpropanoid biosynthesis and pentose and glucuronate interconversions. There were 10,469 DEGs between the 30-mm stage and the 2-mm stage at 15°C, which was much more than the 6242 DEGs between the 30-mm stage and the 2-mm stage at 25°C. At this stage, plant hormone signal transduction and carbon metabolism continued to be regulated, and only up-regulated genes were enriched in the biosynthesis of amino acids and phenylpropanoid biosynthesis; only down-regulated genes were enriched in ribosomes, ribosome biogenesis in eukaryotes, and purine metabolism.
Molecular function enrichment analysis showed that the activities of ATPase, amylase, and abscisic acid 8′-hydroxylase were significantly increased at low temperature during the protrusion stage (Fig. 4). These results indicate that starch is the main substance and energy source for cotton seed germination at the protrusion stage. Transcriptional regulators, peroxidases and oxidoreductases are important participants in low-temperature-tolerant germination and radicle elongation. The results suggest that abscisic acid is the main regulatory hormone for cotton seed germination at low temperature, and seeds require more energy to maintain germination at low temperature.
The comprehensive analysis showed that plant hormone signal transduction and carbon metabolism occurred throughout the entire process of low-temperature seed germination. Regulation of starch and sucrose metabolism occurred only at the 2-mm stage of seed protrusion. Phenylpropanoid biosynthesis was down-regulated at the 2-mm stage and up-regulated when the radicle was 30 mm. Amino acid biosynthesis is initiated at the 30-mm stage of seed germination. This indicates that radicle elongation requires more gene coordination to adapt to radicle growth at low temperature than radicle elongation after protrusion at 15°C. In BP, DEGs were mainly associated with metabolic processes, cellular processes, single-organism processes, and biological regulation. In CC, the DEGs were mainly associated with cells, cell parts, membrane parts, and organelles. In terms of MF, DEGs are mainly associated with binding and catalytic activities. Ge et al. reported that a mutation in the short chain alcohol dehydrogenase gene GhSAD1 resulted in increased cold tolerance in cotton [33]. Peng et al. reported that when seeds are germinated at low temperature, they repair membrane leakage and other damage caused by low temperature with increased antioxidant protective enzyme activity [34]. This experiment also showed that genes related to catalytic activities in seeds exhibited significantly different expressions when the seeds were germinated at different temperatures. The high activity of amylase at low temperature may be one reason why Xinluzao 25 showed low-temperature tolerance.
The KEGG pathway of DEGs showed that 50 different types of metabolic pathways were involved in regulatory changes during the low-temperature germination of Xinluzao 25 (Fig. 6). The proportion of single pathway expressed genes in all expressed genes showed that DEGs were mainly concentrated in starch and sucrose metabolism, phenylpropanoid biosynthesis, and cysteine and methionine metabolism (Fig. 5). Starch and sucrose metabolism (8.84%) and plant hormone signal transduction (6.95%) accounted for the largest proportion of the DEGs. These results indicate that starch and sugar metabolism, phenylpropanoid biosynthesis, and cysteine and methionine metabolism play important roles in the germination of Xinluzao 25 seeds at low temperature. Shen et al. reported that transcriptome analysis of two cotton varieties, cold-tolerant and cold-intolerant, showed that differences in cold-tolerance gene expression during germination were mainly concentrated in energy metabolism, such as glycolysis/gluconeogenesis, the tricarboxylic acid cycle, and the glyoxylate cycle [23]. We concluded that during the process of low-temperature germination in cotton seeds, seeds acquired cold tolerance and low-temperature germination ability by regulating starch and sucrose metabolism.
To survive under adverse conditions, plants activate signal recognition and transmission systems and regulate corresponding enzymes to adapt to the environment. Transcriptome comparison of cold-tolerant and cold-intolerant young cotton leaves and KEGG signaling pathway analysis indicated that plant hormone signal transduction, MAPK signal transduction, and plant–pathogen interactions are involved in low-temperature stress responses [35]. Shu et al. studied the effects of cold stress on the growth of cotton leaves and found that plants adapt to low-temperature environments through the regulation of hormone signaling pathways [36]. Our results showed that hormone regulation of cold-tolerant cotton during low-temperature germination was also an important way for cotton to acquire low-temperature tolerance.
Phenylpropanoids act as bio signals of plant sensitivity to external stimuli and are involved in biological and abiotic stress responses [37]. Widhalm et al. calculated the barrier concentration of the cell membrane in the sphinx flower based on the diffusion model and found that the accumulation of benzene/phenylpropyl or single-sheet VOC on the membrane to a very high concentration without timely transport causes toxicity to the biofilm [38]. Benzene/phenylpropanes can be used as information molecules to regulate the oxidation function of peroxidase through phenylalanine and cinnamic acid [37]. Our results showed that DEGs involved in phenylpropanoid biosynthesis were a large proportion of the total DEGs.
Cysteine and methionine metabolism accounted for the third-largest proportion of DEGs (Fig. 4). Cysteine (Cys) is a semi-essential amino acid and the main component of glutathione (GSH), an important antioxidant in plants. Metabolic regulation of Cys and methionine (Met) can maintain low levels of Cys while ensuring an adequate supply of sulfhydryl groups for essential functions. Antioxidant and free radical elimination is important in plants [39]. Our results suggest that glutathione may be an important protective antioxidant during seed germination of Xinluzao 25 at low temperature.
Previous studies have shown that plant hormones regulate seed germination. ABA and GAs play the most important roles: ABA inhibits seed germination, and GAs promote seed germination [40]. Our results showed that when seeds germinated at a low temperature of 15°C, the gene encoding abscisic acid 8′-hydroxylase was up-regulated, and amylase activity was activated, which promoted the normal germination of seeds at low temperature. The physiological mechanism underlying the cold tolerance of cotton leaves mainly functions by improving SOD and POD activities [35]. Our results showed that the seed-germination ability of Xinluzao 25 was more related to POD enzyme activity and glutathione reductase activity. Amylase, peroxidase, and glutathione reductase were the most active enzymes during low-temperature germination.
The low-temperature-tolerant cotton variety Xinluzao 25 was germinated at 15°C and 25°C, and the transcriptomes of the seed radicles at lengths of 2 and 30 mm at the two temperatures were compared. The mechanism of the gene regulation response of the low-temperature-tolerant cotton variety was discussed. The low-temperature-tolerant variety Xinluzao 25 exhibited much up-regulated and down-regulated gene expression during protrusion and radicle elongation at low temperature. The DEGs were mainly exonic, which accounted for 86.07%–87.63%. In the 2-mm protrusion stage, there were 3521 down-regulated genes and 1289 up-regulated genes in low temperatures; at the 30-mm stage of radicle germination, there were 3811 down-regulated genes and 4159 up-regulated genes when the radicle germinated at low temperature. At the low temperature, the radicle developed from 2 to 30 mm with 8519 up-regulated genes and 4843 down-regulated genes; at normal temperature, there were only 4680 up-regulated genes and 3037 down-regulated genes. By transcriptome analysis, we detected 76,931 genes and 7407 novel genes.
GO classification and KEGG pathway analysis of the DEGs showed that 50 different types of metabolic pathways were involved. The DEGs were mainly associated with three pathways: starch and sucrose metabolism, phenylpropanoid biosynthesis, and cysteine and methionine metabolism. The important mechanism of seed germination cold resistance of Xinluzao 25 is the high activity of starch hydrolase at low temperature. By regulating the phenylpropanoid signaling pathway, the activity of peroxidase was up-regulated, and the reactive oxygen species generated by the low-temperature germination process was eliminated to maintain the stability of the intracellular environment. In addition, glutathione antioxidants were also important antioxidant protective substances during the germination of Xinluzao 25 at low temperature. According to the results of the transcriptome metabolic pathway analysis, the enzyme activities of the two periods were measured. The results showed that amylase, peroxidase, and glutathione reductase were the main enzymes with elevated activities when germination occurred at low temperature.
Acknowledgement: The authors gratefully acknowledge the management of the Henan Institute of Science and Technology, China for laboratory facilities. We thank Candace Webb, Ph.D., from Liwen Bianji (Edanz) (www.liwenbianji.cn), for editing the English text of a draft of this manuscript.
Funding Statement: This research was funded by the Science and Technology Project of Henan Provincial Department of Science and Technology (Item No. 222102110282).
Author Contributions: Study conception and design: Genhai Hu; data collection: Xiuren Zhou, Yuanzhi Fu; analysis and interpretation of results: Genhai Hu, Maoni Chao; draft manuscript preparation: Genhai Hu. All authors reviewed the results and approved the final version of the manuscript.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Warner, D. A., Holaday, A. S., Burke, J. J. (1995). Response of carbon metabolism to night temperature in cotton. Agronomy Journal, 87(6), 1193–1197. https://doi.org/10.2134/agronj1995.00021962008700060026x [Google Scholar] [CrossRef]
2. Pham, H. M., Kebede, H., Ritchie, G., Trolinder, N., Wright, R. J. (2018). Alternative oxidase (AOX) over-expression improves cell expansion and elongation in cotton seedling exposed to cool temperatures. Theoretical and Applied Genetics, 131(11), 2287–2298. https://doi.org/10.1007/s00122-018-3151-1 [Google Scholar] [PubMed] [CrossRef]
3. Lu, X., Jia, X., Niu, J. (2018). The present situation and prospects of cotton industry development in China. Scientia Agricultura Sinica, 51, 26–36. [Google Scholar]
4. Barpete, S., Oguz, M. C., Ozcan, S. F., Anayol, E., Ahmed, H. A. et al. (2015). Effect of temperature on germination, seed vigor index and seedling growth of five turkish cotton (Gossypium hirsutum L.) cultivars. Fresenius Environmental Bulletin, 24(8A), 2561–2566. [Google Scholar]
5. Kargiotidou, A., Deli, D., Galanopoulou, D., Tsaftaris, A., Farmaki, T. (2008). Low temperature and light regulate delta 12 fatty acid desaturases (FAD2) at a transcriptional level in cotton (Gossypium hirsutum L.). Journal of Experimental Botany, 59(8), 2043–2056. https://doi.org/10.1093/jxb/ern065 [Google Scholar] [PubMed] [CrossRef]
6. Yu, Y. W., Yu, G. X., Wei, J. Z. (2019). Analysis of spatial distribution and influencing factors of cotton production lay-out in Xinjiang under the backdrop of supply side reform. Journal of Arid Land Resources & Environment, 33(5), 74–80. [Google Scholar]
7. Cai, L. H., Tai, H. Z., Lian, W. M., Lu, J. B., Dai, C. R. et al. (2021). Effect of sowing time on cotton production in Aral reclamation area. Chinese Cotton, 48(6), 15–18+21. https://doi.org/10.11963/1000-632X.clhdcr.20210611 [Google Scholar] [CrossRef]
8. Wang, G. F., Xu, Q. Q., Wang, A. Q. (2020). The changes of cotton production pattern of whole country and Shandong province in the past two decades and the construction of cotton production protection area supporting system in Shandong Province. Cotton Science, 42(1), 3–13. https://doi.org/10.3969/j.issn.2095-3143.2020.01.001 [Google Scholar] [CrossRef]
9. Rosental, L., Nonogaki, H., Fait, A. (2014). Activation and regulation of primary metabolism during seed germination. Seed Science Research, 24(1), 1–15. https://doi.org/10.1017/S0960258513000391 [Google Scholar] [CrossRef]
10. Yamaguchi-Shinozaki, K., Shinozaki, K. (2006). Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annual Review of Plant Biology, 57(1), 781–803. https://doi.org/10.1146/annurev.arplant.57.032905.105444 [Google Scholar] [PubMed] [CrossRef]
11. Kishitani, S., Watanabe, K., Yasuda, S., Arakawa, K., Takabe, T. (1994). Accumulation of glycine betaine during cold acclimation and freezing tolerance in leaves of winter and spring barley plants. Plant, Cell & Environmen, 17(1), 89–95. [Google Scholar]
12. Jorge, T. F., Rodrigues, J. A., Caldana, C., Schmidt, R., Van Dongen, J. T. et al. (2016). Mass spectrometry-based plant metabolomics: Metabolite responses to abiotic stress. Mass Spectrometry Reviews, 35(5), 620–649. [Google Scholar] [PubMed]
13. Seki, M., Kamei, A., Yamaguchi-Shinozaki, K., Shinozaki, K. (2003). Molecular responses to drought, salinity and frost: Common and different paths for plant protection. Current Opinion in Biotechnology, 14(2), 194–199. https://doi.org/10.1016/S0958-1669(03)00030-2. [Google Scholar] [PubMed] [CrossRef]
14. Wang, D. Z., Jin, Y. N., Ding, X. H., Wang, W. J., Zhai, S. S. et al. (2017). Gene regulation and signal transduction in the ICE-CBF-COR signaling pathway during cold stress in plants. Biochemistry, 82(10), 1103–1117. https://doi.org/10.1134/S0006297917100030 [Google Scholar] [PubMed] [CrossRef]
15. Fowler, S., Thomashow, M. F. (2002). Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. Plant Cell, 14(8), 1675–1690. https://doi.org/10.1105/tpc.003483 [Google Scholar] [PubMed] [CrossRef]
16. Nonogaki, H., Bassel, G. W., Bewley, J. D. (2010). Germination—still a mystery. Plant Science, 179(6), 574–581. https://doi.org/10.1016/j.plantsci.2010.02.010 [Google Scholar] [CrossRef]
17. Ali, G. M. (2014). Effect of temperature regimes on the germination testing of cotton (Gossypium hirsutum L.) germplasm. European Academic, 1(10), 3780–3793. [Google Scholar]
18. Khetran, A. S., Bashir, W., Baloch, S., Saleh, A., Baloch, S. K. (2015). Influence of temperature regimes on germination of cotton (Gossypium hirsutum L.). Varieties, 5(11), 173–177. [Google Scholar]
19. Wei, W., Qi, X., Wang, L., Zhang, Y. X., Hua, W. et al. (2011). Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina pairedend sequencing and development of EST-SSR markers. BMC Genomics, 12, 451. [Google Scholar] [PubMed]
20. Li, X., Hu, H., Hu, X., Wang, G., Du, X. et al. (2022). Transcriptome analysis of near-Isogenic lines provides novel insights into genes associated with seed low-temperature germination ability in maize (Zea mays L.). Plants, 11(7), 887. https://doi.org/10.3390/plants11070887 [Google Scholar] [PubMed] [CrossRef]
21. Wang, X., Wang, L. J., Yan, X. C., Wang, L., Tan, M. L. et al. (2016). Transcriptome analysis of the germinated seeds identifies low-temperature responsive genes involved in germination process in Ricinus communis. Acta Physiologiae Plantarum, 38(1), 1–8. https://doi.org/10.1007/s11738-015-1994-5 [Google Scholar] [CrossRef]
22. Wang, P., Chen, F., Huang, Y., Sun, H., Fan, M. et al. (2021). New insights of low-temperature plasma effects on seeds germination of Platycodon grandiflorum. Israel Journal of Plant Sciences, 68(4), 306–318. [Google Scholar]
23. Shen, Q., Zhang, S., Liu, S., Chen, J., Pang, C. (2020). Comparative transcriptome analysis provides insights into the seed germination in cotton in response to chilling stress. International Journal of Molecular Sciences, 21(6), 2067. https://doi.org/10.3390/ijms21062067 [Google Scholar] [PubMed] [CrossRef]
24. Wang, Y. J., Xie, L., Li, Z. B., Wei, Y. N., Lin, H. R. (2014). Effect of low temperature stress on seed germination and difference evaluation of cold tolerance of cotton in northern xinjiang. Seeds, 33(5), 74–77. https://doi.org/10.3969/j.issn.1001-4705.2014.05.018 [Google Scholar] [CrossRef]
25. Miransari, M., Smith, D. (2014). Plant hormones and seed germination. Environmental and Experimental Botany, 99(3), 110–121. https://doi.org/10.1016/j.envexpbot.2013.11.005 [Google Scholar] [CrossRef]
26. Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Amit, I. (2013). Trinity: Reconstructing a full-length transcriptome without a genome from RNA-seq data. Nature Biotechnology, 29(7), 644–652. https://doi.org/10.1038/nbt.1883 [Google Scholar] [CrossRef]
27. Li, H. S. (2000). Experimental principles and techniques of plant physiology and biochemistry. China: Higher Education Press. [Google Scholar]
28. Anders, S., Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biology, 11(10), 1–28. https://doi.org/10.1186/gb-2010-11-10-r106 [Google Scholar] [PubMed] [CrossRef]
29. Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M. et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Research, 36(1), 480–484. https://doi.org/10.1093/nar/gkm882 [Google Scholar] [PubMed] [CrossRef]
30. Lakkineni, K. C., Bhardwaj, S. N., Abrol, Y. P. (1994). Effect of temperature on early growth and seed-cotton yield in upland cotton (Gossypium hirsutum.L). Indian Journal of Agricultural Sciences, 64(9), 653–654. [Google Scholar]
31. Ozcan, Sancar, Fatih, Sebahattin, Barpete et al. (2015). Effect of temperature on germination, seed vigor index and seedling growth of five turkish cotton (Gossypium hirsutum L.) cultivars. Fresenius Environmental Bulletin, 24(8a), 2561–2566. [Google Scholar]
32. Lukatkin, A. S. (2002). Contribution of oxidative stress to the development of cold-induced damage to leaves of chilling-sensitive plants: 2. The activity of antioxidant enzymes during plant chilling. Russian Journal of Plant Physiology, 49(6), 782–788. https://doi.org/10.1023/A:1020965629243 [Google Scholar] [CrossRef]
33. Ge, C., Wang, L., Yang, Y., Liu, R., Liu, S. et al. (2022). Genome-wide association study identifies variants of GhSAD1 conferring cold tolerance in cotton. Journal of Experimental Botany, 73(7), 2222–2237. [Google Scholar] [PubMed]
34. Peng, M., Chang, Y., Chu, G., Wang, M. (2019). Low-temperature tolerance and transcriptome analyses during seed germination of anabasis aphylla. Journal of Plant Interactions, 14(1), 254–264. https://doi.org/10.1080/17429145.2019.1616840 [Google Scholar] [CrossRef]
35. Cheng, G., Zhang, L., Wang, H., Lu, J., Yu, S. (2020). Transcriptomic profiling of young cotyledons response to chilling stress in two contrasting cotton (Gossypium hirsutum L.) genotypes at the seedling stage. International Journal of Molecular Sciences, 21(14), 5095. https://doi.org/10.3390/ijms21145095 [Google Scholar] [PubMed] [CrossRef]
36. Shu, H., Sun, S., Wang, X., Yang, C., Zhang, G. et al. (2022). Low temperature inhibits the defoliation effificiency of thidiazuron in cotton by regulating plant hormone synthesis and the signaling pathway. International Journal of Molecular Sciences, 23(22), 14208. https://doi.org/10.3390/ijms232214208 [Google Scholar] [PubMed] [CrossRef]
37. Dudareva, N., Klempien, A., Muhlemann, J. K., Kaplan, I. (2013). Biosynthesis, function and metabolic engineering of plant volatile organic compounds. New Phytologist, 198(1), 16–32. [Google Scholar] [PubMed]
38. Widhalm, J. R., Jaini, R., Morgan, J. A., Dudareva, N. (2015). Rethinking how volatiles are released from plant cells. Trends in Plant Science, 20(9), 545–550. https://doi.org/10.1016/j.tplants.2015.06.009 [Google Scholar] [PubMed] [CrossRef]
39. Tang, W. J., Tu, Q., Liu, Z. Q., Huang, R. L. (2010). Research progress of cysteine metabolism andimmune function. Life Sciences, 22(2), 150–154. https://doi.org/10.13376/j.cbls/2010.02.014 [Google Scholar] [CrossRef]
40. Guo, L. H., Chen, S. N., Gong, M. (1999). Comparison of methods for determination of glutathione reductase activity in maize. Journal of Yunnan University (Natural Science Edition), 21(5), 416–418. https://doi.org/10.3321/j.issn:0258-7971.1999.05.024 [Google Scholar] [CrossRef]
Supplementary Materials
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools