
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Phospholipid Metabolism Reprogramming of Cancer Stem Cells and Its Impacts on Stemness
State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, Research Unit of Oral Carcinogenesis and Management, Chinese Academy of Medical Sciences, West China Hospital of Stomatology, Sichuan University, Chengdu, 610041, China
* Corresponding Authors: Yu Zhou. Email: ; Yingqiang Shen. Email:
(This article belongs to the Special Issue: Stem Cells Therapy in Health and Disease)
BIOCELL 2025, 49(4), 579-605. https://doi.org/10.32604/biocell.2025.060045
Received 22 October 2024; Accepted 31 January 2025; Issue published 30 April 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Cancer Stem Cells (CSCs) are cancer cells with self-renewal and tumorigenesis abilities. CSCs in tumor tissues are the leading cause of tumor progression, recurrence, and drug resistance. CSCs have distinct metabolic features that contribute to maintaining their self-renewal and stemness. Phospholipids are essential components of cell membranes and play fundamental roles in cellular activities. CSCs have abnormal phospholipid metabolism, which affects their self-renewal, differentiation, invasion, and drug resistance. Compared with non-CSCs, the phospholipid metabolism of CSCs mainly focused on significantly increased fatty acid (FAs) and phospholipids synthesis, phospholipid unsaturation, and lipolysis-fatty acid oxidation (FAO). In brief, FA and phospholipid metabolism in the anabolic and catabolic pathways are strictly regulated in CSCs to maintain self-renewal and stemness activity. In this review, we summarize the alterations in phospholipid metabolism in CSCs and their impacts on the stemness of CSCs, and we put forward the potential applications of targeting phospholipid metabolism for CSCs, to provide directions for the development of drugs targeting the phospholipid metabolism.Graphic Abstract
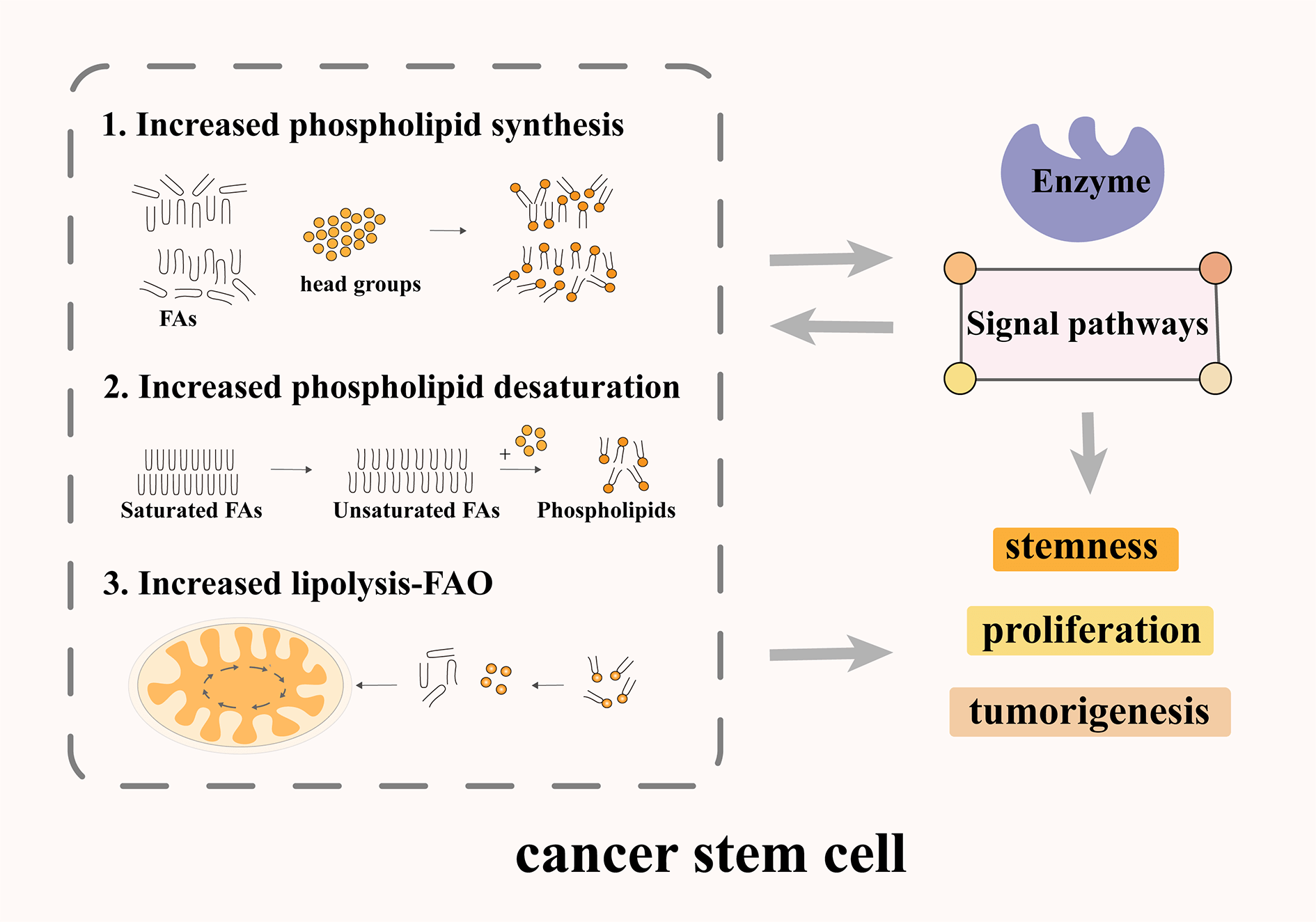
Keywords
Highlights
1- Increased phospholipid synthesis, desaturation, and FAO in CSCs.
2- Effect of phospholipid metabolizing enzymes on the characteristics of CSCs.
3- Effect of phospholipid regulated signaling pathway on the characteristics of CSCs.
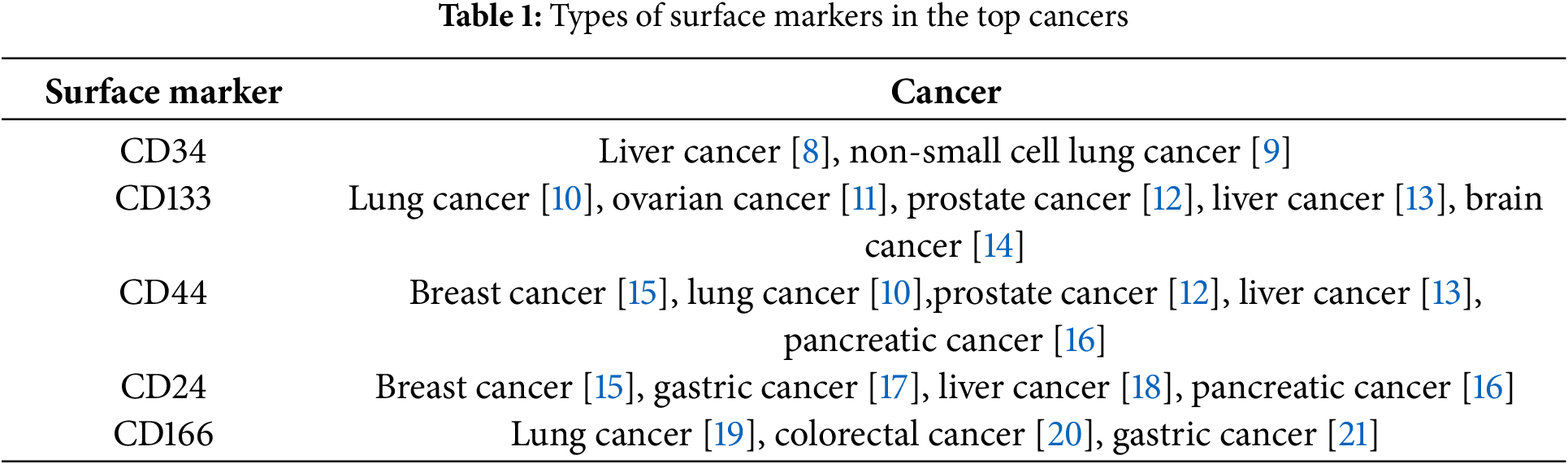
Since the discovery of CSCs in acute myeloid leukemia, researchers have identified CSC-like cells in a variety of solid tumors, including lung, breast, and pancreatic cancer [1–4]. These CSCs represent a distinct subset of tumor cells with the ability to self-renew, playing a crucial role in tumor initiation and progression, and significantly influencing the prognosis of cancer patients [5]. CSCs exhibit several important biological characteristics (Fig. 1). First, like normal stem cells, CSCs possess the ability to self-renew and can generate more CSCs through division, thereby maintaining tumor growth and expansion. This ability enables CSCs to survive treatments and plays a crucial role in tumor recurrence. Second, CSCs have differentiation potential. Although they can self-renew, they can also differentiate into various tumor cell types. This contributes to tumor heterogeneity, which complicates treatment. Third, CSCs have strong tumor-forming capabilities. In a suitable microenvironment, they can give rise to new tumors, making them a source of tumor recurrence and metastasis. Fourth, CSCs generally exhibit high drug resistance. They can evade chemotherapy and radiotherapy through mechanisms such as the activation of anti-apoptotic pathways. This resistance is a key factor in the failure and recurrence of cancer treatments. These biological characteristics make CSCs a critical focus in tumor biology research and cancer treatment. Understanding these features is essential for developing new therapeutic strategies, particularly targeting CSCs. In, addition, a defining feature of CSCs is their ability to generate phenotypically and functionally diverse cell populations. Surface hallmarks, biomolecules highly expressed on the cell membrane of CSCs, play a crucial role in stem cell maintenance and tumorigenicity. Although these hallmarks vary across solid tumors and only a few common markers exist between them, they can be utilized for CSC isolation, characterization, and targeted therapy [6,7]. Ongoing research continues to uncover potential surface hallmarks of CSCs, Table 1 summarizes the biomarker molecules of CSCs.
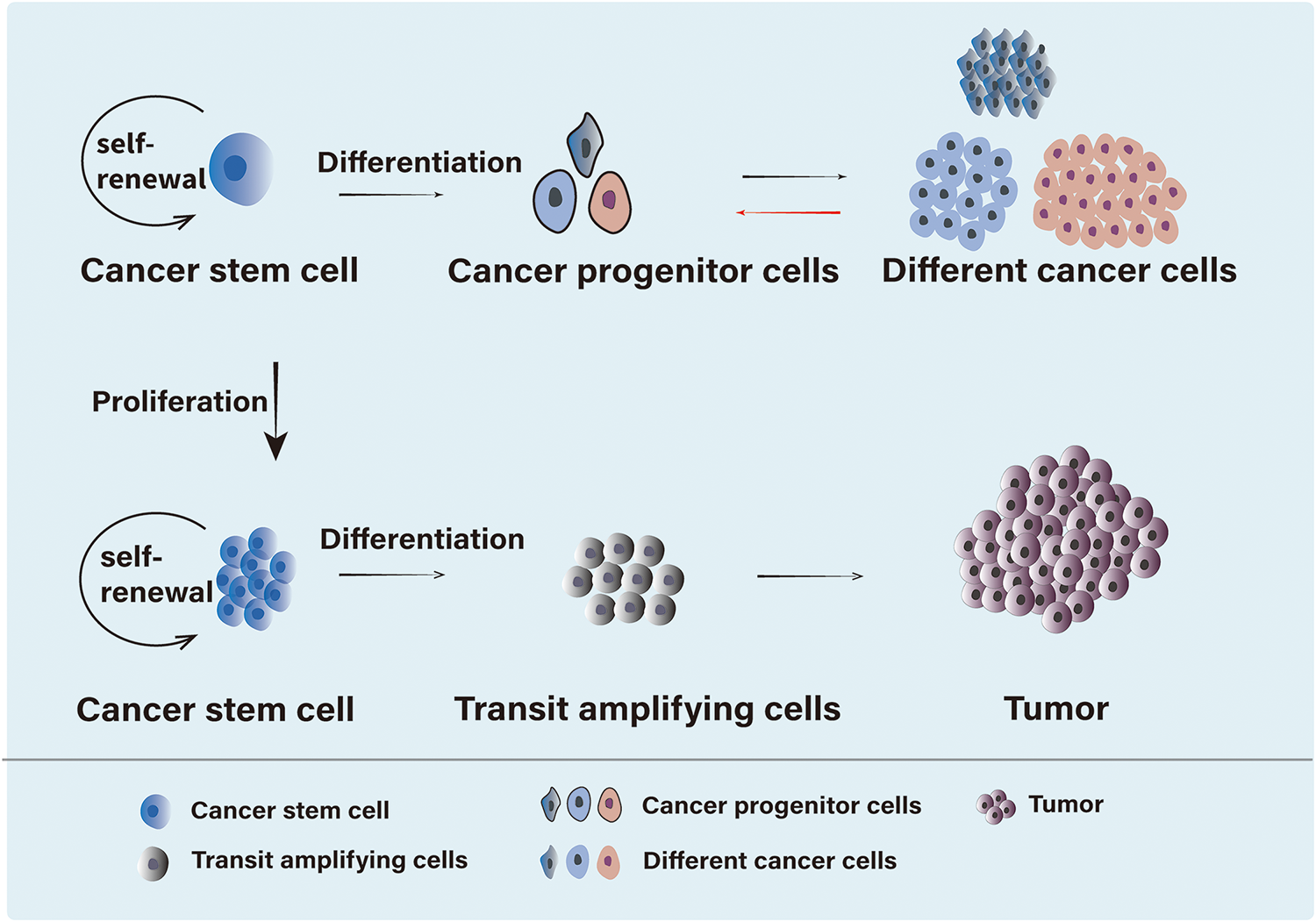
Figure 1: Differentiation of CSCs. CSCs are a type of cell with self-renewal, differentiation potential, and tumor-initiating ability. On one hand, CSCs can differentiate into cancer progenitor cells, which then give rise to heterogeneous cancer cells, or into transit amplifying cells, ultimately leading to tumor formation. On the other hand, CSCs possess self-renewal capability, allowing them to maintain a subset of daughter cells with the same self-renewal potential while differentiating, thus preserving the stability of the CSC population
Phospholipids are crucial components of cell membranes, contributing to both structural diversity and energy storage, as well as supporting various metabolic processes. They play key roles in regulating cellular functions, including cell adhesion, migration, signal transduction, and apoptosis [22]. The metabolic intermediates and by-products of phospholipids are involved in numerous cellular activities, supplying energy and modulating signaling pathways that influence cell morphology and function [23]. Furthermore, enzymes responsible for phospholipid metabolism help regulate essential biological processes, such as the cell cycle and cell proliferation [24].
The metabolism of nutrients in the tumor microenvironment undergoes dynamic changes throughout tumor progression. Recent clinical and experimental evidence indicates that phospholipid metabolism plays a role in tumorigenesis, particularly in critical oncogenic signaling pathways [25,26]. Notably, phospholipid metabolism is reprogrammed in CSCs, and these alterations regulate the phenotype of CSCs to some extent, such as the expression and maintenance of stemness, invasion, and metastasis [27]. Studies have identified certain phospholipid metabolites and enzymes as potential metabolic markers for CSCs [28]. In brief, phospholipid metabolism maintains rapid tumor proliferation, invasion, and metastasis [29,30]. This review examines the metabolic changes in phospholipids in CSCs and their regulatory effects on CSC stemness.
2 Classification, Synthesis, and Metabolism of Phospholipids
Phospholipids are lipids that contain phosphoric acid and primarily include glycerophospholipids and sphingomyelin. As key components of cell membranes, phospholipids exhibit a diverse range of chemical structures and functions. Phospholipid metabolism encompasses a complex network of biochemical processes, regulated by various enzymes and transporters. These processes include the synthesis, transformation, and degradation of phospholipids, as well as the synthesis, uptake, transport, and breakdown of glycerol and FA (Fig. 2).
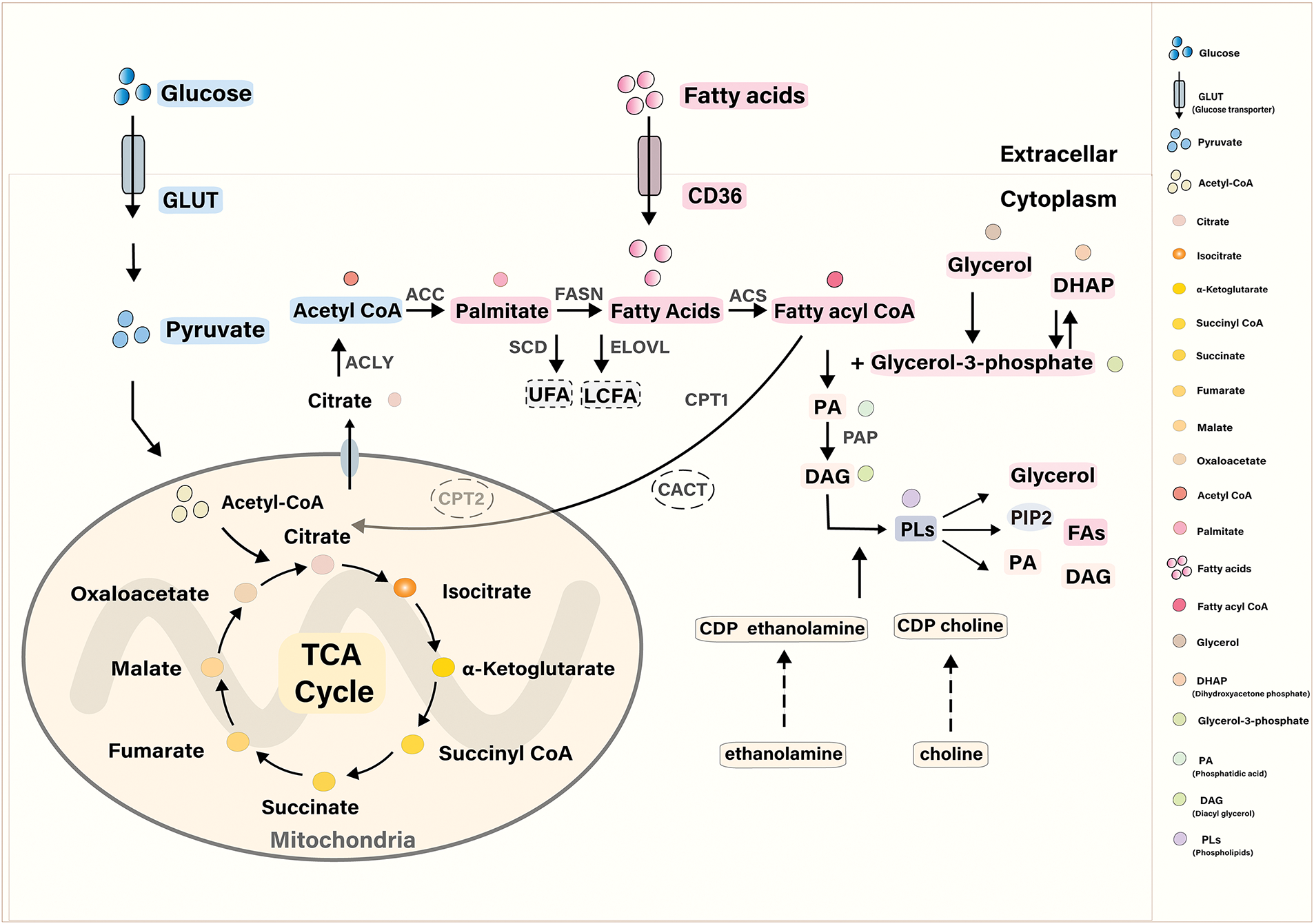
Figure 2: Synthesis, decomposition, and transformation of Phospholipids. In de novo phospholipid synthesis, FAs are absorbed by FABP and CD36, converted into acyl-CoA by ACS, and then react with glycerol-3-phosphate to form PA. Glycerol-3-phosphate is generated either by glycerol kinase from glycerol or by glycerol-3-phosphate dehydrogenase from DHAP. FAs promote phospholipid synthesis by esterifying with glycerol-3-phosphate to form PA. FAs can be derived from glucose. Glucose enters the cell via GLUT transporters, is converted into pyruvate, and then into acetyl-CoA in the mitochondria, where it participates in the citric acid cycle. Citrate is enzymatically converted into palmitate and FAs by enzymes such as ACLY, ACC, and FASN. In the endoplasmic reticulum, palmitate can be converted into UFAs by SCD or elongated into LCFAs by ELOVL. Phospholipids can then be converted into various products, including glycerol, PA, diacylglycerol, and others, through the action of different enzymes
The basic structure of glycerophospholipids consists of a glycerol backbone, FAs, phosphoric acid, and a group attached to the phosphoric acid. Based on the nature of the attached group, glycerophospholipids are primarily classified into phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI), phosphatidic acid (PA), and cardiolipin (CL). Glycerophospholipids are synthesized in all tissue cells, primarily through the diglyceride and cytidine diphosphate (CDP)-diglyceride pathways. FAs are essential precursors in the synthesis of phospholipids. Citrate is converted to palmitate and Fas through a series of enzymatic reactions such as ATP citrate lyase (ACLY), acetyl CoA carboxylase (ACC), and fatty acid synthase (FASN). In the endoplasmic reticulum, palmitate can be converted to unsaturated FAs (UFAs) by stearoyl COA desaturase (SCD) or further extended by elongases of very long-chain fatty acids (ELOVL) to generate long-chain FAs (LCFA) [31–33]. During the de novo synthesis of phospholipids, FAs are taken up as primary precursors via fatty acid binding protein (FABP) and the fatty acid transporter CD36. FA is converted to fatty acyl-CoA under the action of fatty acyl-CoA synthetase (ACS) and reacts with glycerol-3-phosphate to generate PA. Glycerol-3-phosphate has two primary sources: it can be generated from glycerol by the enzyme glycerol kinase, or from dihydroxyacetone phosphate (DHAP) by glycerol-3-phosphate dehydrogenase. Additionally, studies have shown that FAs contribute to phospholipid synthesis by esterifying with glycerol-3-phosphate to form PA [34]. PA generates diglyceride (DAG) under the action of phosphatidic acid phosphatase (PAP), further generating PC, PE, and other phospholipids. Phospholipids can also undergo conversion via intermolecular head group exchange, such as when ethanolamine is substituted for serine. Additionally, due to the structural diversity of phospholipids, the composition of fatty acyl chains in mammalian cells is also regulated by deacylation and reactive acylation, a pathway referred to as the Land’s cycle [35]. Sphingomyelin, commonly found in brain tissue, consists of sphingosine, FAs, and various head groups (including phosphocholine and phosphoethanolamine. Sphingomyelin synthesized from phosphocholine is the most common and essential component of various membrane structures of neural tissues [36].
Phospholipases catalyze the breakdown of phospholipids, generating glycerol, FAs, and other bioactive substances such as lysophospholipids, ceramide, and sphingosine. Glycerol can be converted into glycerol-3-phosphate, which is used for phospholipid synthesis, and gluconeogenesis to produce glucose, or converted into DHAP, an intermediate in glycolysis. DHAP can then enter the glycolytic pathway and the tricarboxylic acid (TCA) cycle. Fs can be reused for phospholipid synthesis or enter the final stage of fatty acid metabolism—FAO, also known as β-oxidation. First, long-chain acyl CoA synthetases (ACSLS) activate free FAs and convert them into fatty acyl-CoAs. The transfer of fatty acyl-CoAs through the mitochondrial membrane is a critical rate-limiting step. Acyl-CoAs are converted into fatty acylcarnitines by carnitine palmitoyltransferase 1 (CPT1) in the presence of carnitine, and the fatty acylcarnitines are then transported into mitochondria by carnitine acylcarnitine translocase (CACT). Fatty acylcarnitines are eventually reconverted to fatty acyl-CoA by CPT2. Fatty acyl-CoAs enter the mitochondrial matrix and undergo β-oxidation, catalyzed by the oxidase system. This process involves four sequential reactions: dehydrogenation, hydration, another dehydrogenation, and thiolysis. The result is the formation of acetyl-CoA and a shortened fatty acyl-CoA, which has two fewer carbon atoms than the original. Acetyl-CoA enters the TCA cycle. Phospholipid metabolism can also involve many bioactive substances. Sphingosine is converted to sphingosine 1 phosphate (S1P) under the action of sphingosine kinase (SPHK), with lysophosphatidic acid (LPA), ceramide, and other mediators acting as second messengers to regulate cellular activities, resulting in a variety of biological effects.
3 Phospholipid Metabolic Reprogramming in CSC
Due to the rapid proliferation of tumor cells and insufficient angiogenesis of tissues, tumors usually live in hypoxic, acidic, and nutrient-deprived microenvironments. Therefore, tumor cells must regulate their cellular bioenergetics effectively to cope with the adverse environment. This process is called metabolic reprogramming [37,38]. CSCs showed prominent metabolic characteristics. More and more studies have shown that metabolic reprogramming and CSC characteristics are two highly intertwined processes in tumor development [39]. In recent years, studies have shown that reprogramming of phospholipid metabolism is closely related to the occurrence, progression, and prognosis of tumors, especially CSCs, including ovarian CSCs, breast CSCs, endometrial CSCs, pancreatic cancer stem cells (PaCSCs) and glioma stem cells (GSCs), etc. Phospholipid metabolism has similar characteristics in CSCs or CSC-like cells of these different tissues and organs. For example, there are different degrees of increase in phospholipid content and FAO in breast CSCs and ovarian CSCs, and the degree of phospholipid unsaturation and FAO metabolism are significantly increased in gastric cancer stem cells (GCSCs) and GSCs.
3.1 Increased Phospholipid Synthesis
In tumor tissue, both endogenous and exogenous lipids contribute to a significant increase in FA and phospholipid content due to the high metabolic demand in CSCs, where the synthesis rate of these lipids accelerates [40]. CSCs are heterogeneous populations with different metabolic states associated with their potential for tumor initiation. Quiescent CSCs have low metabolic rate and slow cell division [41]. Enzymes involved in FA and phospholipid synthesis, including FASN, SCD, PAP, etc., are significantly upregulated in CSCs [42–45]. It has been demonstrated that phospholipid content increases with cell transformation and tumor progression, regulating various oncogenic processes, such as tumor growth, CSC formation, and metastasis. For example, lipid analysis of breast cancer tissue demonstrated a significant increase in phospholipid content. PA and its product, LPA, are increased in cancer cells and CSCs compared to nonmalignant cells. Seo et al. isolated spherical cells A2780-SP from A2780 epithelial ovarian cancers (EOCs) and determined their CSC-like properties by identifying their high sphere-forming ability, drug resistance to chemotherapy such as paclitaxel or cisplatin, and expression of stem cell markers including SRY-box transcription factor 2 (SOX2) and aldehyde dehydrogenase 1 (ALDH1) [46]. Their results demonstrated that the ATX-LPA-LPAR1 signaling axis was involved in maintaining ovarian CSC stemness, and ovarian CSCs could enhance their stem cell properties when treated with LPA. This included inducing stem cell-related gene expression, enhancing microsphere formation, increasing chemotherapy resistance, and boosting tumor-initiating potential in mice. CSCs can also utilize the metabolic intermediates from the Warburg effect to synthesize phospholipids, further enhancing their self-renewal growth properties. Lysophosphatidylcholine acyltransferase (LPCAT), a vital enzyme of the Land’s cycle, is involved in tumor progression through membrane phospholipid remodeling [47]. A recent study found that overexpression of LPCAT1 alters the contents of PC and PE, leading to increased stemness and metastasis of CSCs [48].
By comparing CSCs with non-stem cells, researchers found that CSCs contain more UFAs and phospholipids than non-stem cells, affecting the stemness of cancer cells and the formation of CSCs [49]. The metabonomic results of a colon cancer study also indicated that CSCs contained higher levels of unsaturated phospholipids, with similar effects on CSCs [50]. In GSCs, researchers found that the synthesis and utilization of polyunsaturated FAs (PUFAs) were significantly increased, thereby changing the composition and structure of membrane phospholipids [51]. Unsaturated lipid is considered a unique metabolic biomarker of ovarian CSCs [52]. ALDH and CD133 are described as characteristic markers of CSCs. The authors used hyperspectral stimulated Raman scattering (SRS) microscopy to chemically image ALDH+/CD133+ cells to identify and characterize the unsaturation of lipids in ovarian CSCs. It is known that the peak at 3002 cm is mainly from unsaturated lipids, while the lipid droplets in ALDH+/CD133+ cells have a stronger signal at 3002 cm, which means that ALDH+/CD133+ CSCs have a higher level of unsaturated lipids. Stearoyl-CoA desaturase 1 (SCD1) first attracted attention for its role in the pathway that produces monounsaturated fatty acids (MUFAs). Functionally, SCD1 is a rate-limiting enzyme that catalyzes the formation of MUFAs from saturated FAs. Most of the UFAs in the body are products of SCD1 catalysis. Moreover, SCD1 is highly expressed in liver, lung, breast, and other tumors and shows an increased proportion of unsaturated lipids. In recent years, studies have found that SCD1 is closely related to CSCs. Overexpression of SCD1 promotes the proliferation and migration of CSCs and inhibits cell death [53]. Blocking SCD1 reduces the expression of ovarian CSC markers and prevents tumorigenesis in vivo, which is consistent with the role of SCD1 in breast cancer [54]. Furthermore, SCD1 was overexpressed in primary GCSCs, and SCD1 inhibition reversed the epithelial-mesenchymal transformation (EMT) process and reduced the probability of gastric cancer metastasis in vitro and in vivo [55].
A new pathway of lipid desaturation remodeling has recently been identified and shown to regulate the final effects of SCD1 [56]. Sapienate and another fatty acid desaturase 2 (FADS2) were discovered. They also found that when SCD1 is inhibited, FADS2 compensates for the loss of SCD1 activity. Elevated FA unsaturation is positively correlated with SCD1 or FADS2 levels and the oncogenic capacity of ovarian cancer cells. Inhibition of SCD1/FADS2 retards tumor growth and reduces CSC formation and decreases drug resistance. Phospholipid desaturation in CSCs was further investigated, potentially opening new opportunities to target them.
The conversion and utilization of phospholipid metabolites such as FAs, glycerol, and choline by CSCs is increased in different tumors. Many types of tumors use FAO as their primary energy source [57]. Studies have shown that triple-negative breast cancer (TNBC) controls tumor growth via FAO and promotes metastasis. FAO also activates Src oncoprotein in TNBC, associated with acquiring tumor cell stemness [58]. Inhibiting FAO decreases the self-renewal capacity and tumorigenicity of breast and liver CSCs while increasing their sensitivity to chemotherapy [59,60]. Therefore, increased FAO in CSCs is critical for CSC generation and survival in response to inflammatory signaling. FABP plays a vital role in FA uptake and transports FAs to different organelles to function after uptake. A recent study found that FABP promotes TNBC progression and breast CSCs activity. In vitro and in vivo studies have shown that knocking down FABP4 inhibits breast CSC activity, such as tumor-initiating ability. In vivo studies have also shown that knocking down FABP4 reduces metastatic ability and tumor progression in TNBC [61]. A recent study found that FAO activity increased in PaCSCs, with related metabolic genes also upregulated. Active metabolism may contribute to the metastasis of PaCSCs. In addition, exogenous supplementation of FAs significantly enhanced the self-renewal capacity and tumor formation abilities of CSCs. Inhibiting FAO metabolism has the opposite effect, reducing the self-renewal and tumor-forming abilities of CSCs, while enhancing their sensitivity to chemotherapeutic drugs [62] Leukemia stem cells (LSCs) increase free FAs by secreting pro-inflammatory factors and promoting lipolysis. These FAs enter LSCs via CD36 and are eventually utilized for energy production through FAO to support stem cell survival and evade chemotherapy [63]. CD36 expression is elevated in GSCs, and the uptake of oxidized phospholipids promotes GSC proliferation [64]. Furthermore, Singh et al. found that the coat protein complex I (COPI)-Arf1 complex regulates stem cell survival through the lipolysis FAO pathway. The knockdown of the associated genes blocks lipolysis and selectively kills stem cells. They also used an ARF1 inhibitor and found that CSC formation was significantly reduced in Drosophila but had a negligible effect on normal stem cells, which was somewhat sufficient to visualize the dependence of CSCs on the lipolysis-FAO [65].
In addition, FAO produces abundant antioxidants, such as NADPH, which can promote the survival of CSCs. FAO reduces the production of reactive oxygen species (ROS) that are detrimental to stem cells. However, studies have also shown that noncytotoxic doses of ROS can instead promote neoplastic transformation and stem cell features in renal epithelial cells, which may explain the increase in the content and utilization of unsaturated phospholipids and UFAs in CSCs, suggesting that an appropriate amount of ROS is required to maintain stemness [66].
4 Altered Enzymatic Activity in Phospholipid Reprogramming in CSCs
Enzymes involved in phospholipid metabolism have been evaluated in different cancers, and the results showed that they are involved in tumorigenesis and development, especially in cell carcinogenesis, tumor progression, and cancer cell properties (such as rapid proliferation, migration, and invasion) and have important regulatory roles. These enzymes include choline kinase α (CHKα), ethanolamine kinase (ETNK), phospholipase A (PLA), phospholipase D (PLD), phospholipase C (PLC), sphingomyelinase (SM), among others [67].
CHKα is a critical enzyme in the CDP-choline pathway, involved in the first step of lecithin synthesis, and phosphorylates choline to phosphorylcholine to form phosphorylcholine, which is essential for cell membrane structures. Its encoding gene is an oncogene that promotes tumorigenesis and progression. In breast, ovarian, colorectal, and other cancers, gene overexpression and enzyme activity highly increased, resulting in higher levels of phosphorylated choline in tumor tissue [68–70]. CHKα can regulate cell proliferation and tumorigenesis in breast and prostate cancers [71]. CHKα can also induce lipid droplet degradation when energy is insufficient [72]. MN58b, a derivative of HC-3, a small molecule inhibitor of CHKα, has been shown to inhibit CHKα activity. C/EBP homologous protein (CHOP), a major pro-apoptotic transcription factor induced by endoplasmic reticulum stress, is activated by CHKα inhibition, which exacerbates the stress response in cancer cells and triggers apoptosis through this pathway [73]. In addition, Koch et al. found that recurrence and metastasis of glioblastoma were mainly driven by EMT and brain CSCs. Inhibition of CHKα regulating choline metabolism can significantly reduce the expression of EMT-related genes in glioblastoma cells and invasiveness [74]. Lacal et al. found that the combination of traditional chemotherapeutic agents and choline kinase inhibitors improved survival and quality of life in patients with non-small cell lung cancer (NSCLC), suggesting that inhibition of CHKα may be the basis for the development of new treatments [75].
PLD is a hydrolase enzyme that acts on lecithin, producing PA and choline [76]. PA functions as a second messenger that binds directly to the mammalian target of Rapamycin (mTOR) and can also be converted into DAG via PAP or into LPA via PLA2. Studies have confirmed that PLD directly affects proliferation, invasion, metastasis, and drug resistance [77]. Elevated PLD activity or increased expression of PLD1 and PLD2 has been observed in various cancers and transformed cells. Activated PLD promotes the production of PA, which in turn activates Yes-associated protein (YAP) signaling, contributing to drug resistance in squamous cell carcinoma [78]. Muñoz-Galván et al. found that PLD2 is overexpressed in colon cancer, and its product, PA, induces senescence-associated secretory phenotype (SASP) that increases the stem cell properties. They also confirmed this link in a mouse experimental model, showing that high PLD2 expression increases stemness and tumorigenesis [79]. More recently, they reported that PLD2 is also overexpressed in ovarian cancer, contributing to chemoresistance. They found that hypoxia-inducible factor 1 (HIF-1) is required for maintaining CSCs, and hypoxia-induced ovarian cancer stemness depends on PLD2 expression. In summary, the HIF-1α-PLD2 axis promotes the generation and maintenance of CSCs under hypoxic conditions [80] Upregulation of PLD has been linked to increased occurrence and invasion of breast tumors and bladder cancer [81,82]. A study found that PLD1 is elevated in GSCs, especially in recurrent glioblastoma, and this upregulation correlates with GSC resistance to temozolomide. Inhibition of PLD1 sensitizes GSCs to temozolomide and reduces the occurrence of glioblastoma [83]. Additionally, a recent study revealed that PLD1 regulates the self-renewal capacity of cancer-initiating cells through the E2F1–miR-4496–β-catenin axis. Importantly, PLD1 inhibition specifically targets GSCs, not neural progenitor cells, and reduces the self-renewal capacity of CSCs in colorectal cancer and gastric cancer. Therefore, PLD1 may serve as a novel therapeutic target for GSCs [84]. In addition, PLD has been positively associated with rapamycin resistance in human breast cancer cells [85].
PLD also links survival and migration signals of human cancer cells to stress responses. Interestingly, PLD does not always act independently within cells. PLD1 and CHKα interact. CHKα silencing increases PLD1 expression and vice versa [86]. Therefore, whether alone or in potential synergy, PLD is undoubtedly a promising therapeutic target for cancer treatment [87].
PLC cleaves the phosphate group from phospholipids, and it exists in two main types: phosphatidylcholine-specific and phosphatidylinositol-specific [67]. The former hydrolyzes PC to generate DAG and phosphorylcholine, while the latter hydrolyzes phosphatidylinositol-4,5-bisphosphate (PIP2) to produce DAG and IP3, both of which are the key second messengers in cells. PC is the most abundant in cells, while PI, especially PIP2, only exists in trace amounts. Based on this, phosphatidylcholine-specific PLC may produce more persistent DAG elevation than phosphatidylinositol-specific PLC [67]. DAG activates protein kinase C and induces sustained mitotic signaling, possibly leading to cellular transformation. Correlative studies have demonstrated that phosphatidylcholine-specific PLC activity and DAG levels are elevated, and cell transformation and loss of contact inhibition occur in ovarian [88], breast [89], and squamous cell carcinomas [90]. Among them, ovarian cancer studies have shown that PLC is related to cancer cell transformation, proliferation, migration, and invasion. Squamous cell carcinomas showed differential stemness potential with up-regulated mRNA levels of stemness-related markers, which suggests that PLC is associated with transforming non-stem cells to stem cells in tumor tissues. Research by Schnoeder et al. found that Phospholipase C Gamma 1 (PLCγ1) is highly expressed in hematopoietic stem cells and myeloid leukemia and concluded that PLCγ1 is required for the maintenance of LSCs [91]. Furthermore, Okada et al. indicated that PLCε plays a crucial role in maintaining the survival, stemness, and tumor-initiating ability of GSCs [92].
SM acts similarly to PLC by releasing phosphocholine or phosphoryl ethanolamine from sphingomyelin to generate ceramide. In addition, cancer cells can convert ceramide into sphingosine, which is subsequently phosphorylated by sphingosine kinase1 (SK1) to produce S1P. S1P also belongs to lysophosphatide and is an essential bioactive mediator in the cellular process. Recent studies have shown that SM and SK1 are involved in biological processes such as cell cycle arrest, migration, and inflammation and the promotion of breast cancer development through regulation of CSCs apoptosis and proliferation [93]. SM exerts its anti-cancer effects by releasing ceramide and can be divided into three types based on its optimal pH: acidic sphingomyelinase (ASM), neutral sphingomyelinase (nSMase), and alkaline sphingomyelinase (alk-SM) [94,95]. Studies have shown that cells or mice deficient in ASM resist apoptosis induced by anti-tumor stimuli. Conversely, overexpression of ASM sensitizes cancer cells to chemotherapy and radiotherapy [96]. Drug-induced ASM-ceramide has been shown to improve microvascular function, making radiation-resistant colorectal CSCs more responsive to radiotherapy [97]. Therefore, ASM is a potential target for anticancer therapy. nSMase inhibits tumor growth and promotes apoptosis. In the nSMase-2 deficient mice, the formation of liver tumors was increased, and the tumor tissue significantly expressed CD133 and EpCAM mRNA levels, which were markers of liver cancer CSCs [98]. CD133+ cells exhibited increased sphingomyelin and ceramide levels, suggesting that the absence of nSMase-2 supports the survival and proliferation of CSCs. Lw et al. showed that overexpression of SK1 can enhance the survival and proliferation of breast CSCs. They identified that signal transducer and activator of transcription 1 (STAT1) and Interferon (IFN) signal are the critical regulatory targets of SK1, with SK1 activation suppressing STAT1 signaling [99]. Moreover, SK1 overexpression enhances the stemness and self-renewal of ovarian cancer cells through SOX2-dependent mechanisms [100]. In addition, it has long been reported that S1P and its receptor S1PR play an essential role in promoting the proliferation and dry phenotype of CSCs [101]. SK1 promotes growth and inhibits apoptosis. S1P, produced by SK1, can be secreted through specific transporters, acting through autocrine or paracrine. Different S1P receptors are coupled with distinct G proteins to regulate various signaling pathways, including mitogen-activated protein kinase (MAPK), phosphatidylinositol-3-kinase/protein kinase B (PI3K/AKT), and PLC-mediated signaling pathways [102].
In addition to the above enzymes, PLA2 has also been implicated in regulating CSCs and maintaining stemness [103]. PLA2 catalyzes the release of FAs from the second carbon group of glycerol. As an extracellular pattern of PLA2, secreted phospholipase A2 (sPLA2) is widely involved in different types of human cancers. Long non-coding RNA (LncRNA) plays a vital role in biological processes such as cell metabolism and stemness maintenance [104]. Xu et al. found that sPLA2 overexpression promoted cell migration, invasion, and stemness [105]. Silencing LncRNA SLNCR1 downregulated sPLA2 expression, while sPLA2 overexpression can weaken the inhibitory effect of LncRNA SLNCR1 silencing on cell stemness. This indicates that LncRNA SLNCR1 may regulate the stemness in NSCLC by interacting with sPLA2. PLA2G16, is a member of the current PLA2 family. Liu et al. found a new LncRNA regulating phospholipid metabolism in BCSCs by bioinformatics analysis called LncROPM [106]. They discovered that in BCSCs LncROPM significantly promoted phospholipid metabolism and free FA production by increasing the stability of corresponding mRNA, especially arachidonic acid which activated signaling pathways such as PI3K/AKT and Hippo/YAP, which participated in the maintenance of BCSC stemness. Notably, the study found that LncROPM-PLA2G16 signaling significantly enhanced the chemoresistance of BCSCs, suggesting that targeting the LncROPM-PLA2G16 axis may be a new treatment strategy for breast cancer patients.
5 Phospholipids Affect CSCs by Modulating Signal Transduction
In addition to participating in energy metabolism and cell membrane formation, phospholipids also participate in signal transduction, where they help regulate various cellular functions [107]. In phospholipid metabolism, several messengers activate tumor-related signal pathways, such as PI3K/AKT, Ras/ERK, and MAPK pathways [108]. Phospholipid metabolism is closely linked to the carcinogenic signal pathway (Fig. 3). For example, it affects the cell membrane by changing phospholipid molecules’ structure, composition, and content, thus regulating the clustering of membrane receptors and modulating downstream carcinogenic signaling [109]. Many studies have shown that phospholipids and their metabolites play a crucial role in maintaining the phenotype of cancers. They promote tumor proliferation, enhance drug resistance, and induce stemness-related gene expression, all of which are closely related to abnormal activation of signal pathways [110,111].
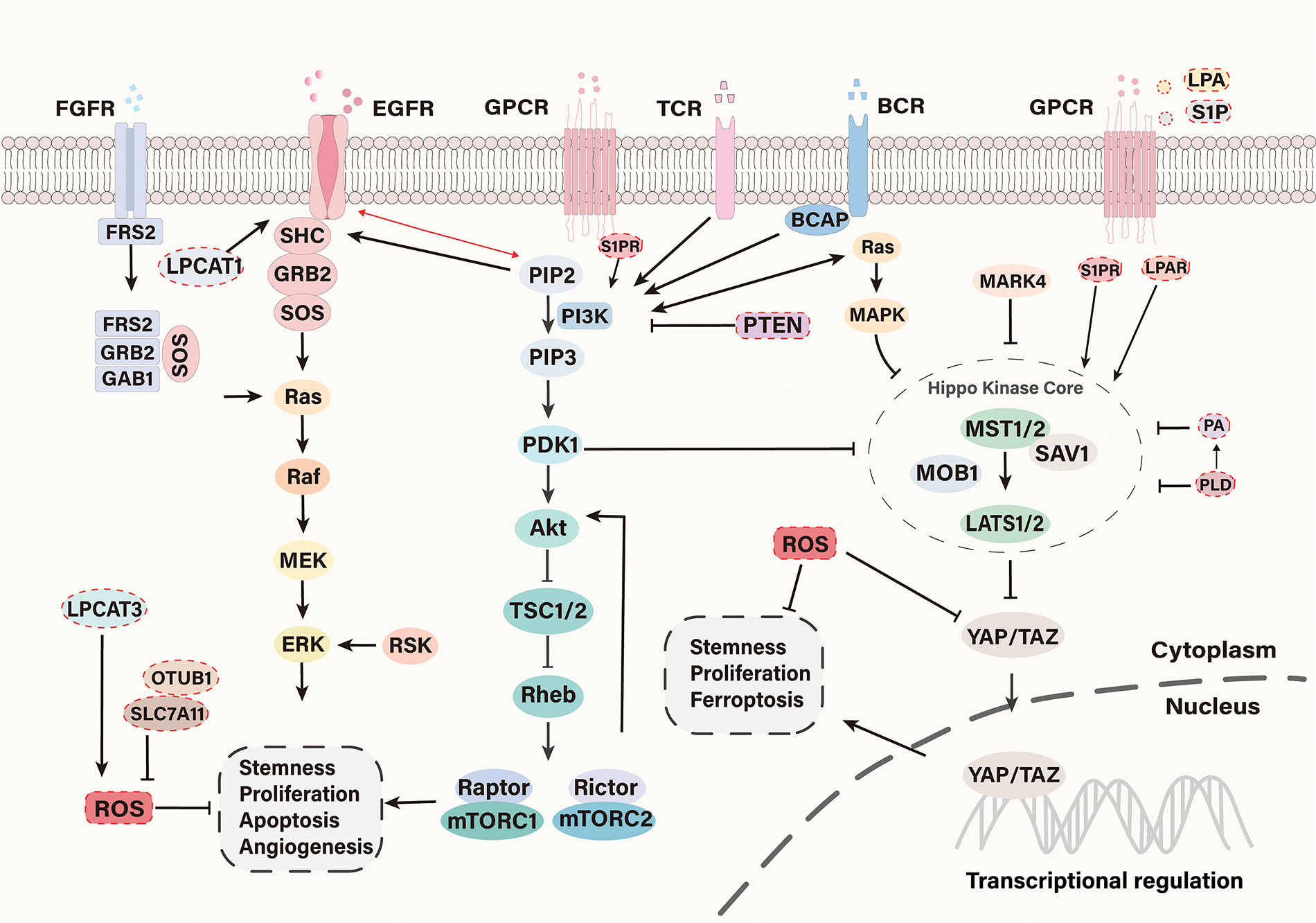
Figure 3: Signal pathways involved in CSC regulation. In the phospholipid metabolism of CSCs, tumor-associated signaling pathways such as PI3K/AKT, RAS/ERK, and Hippo/YAP are often aberrantly activated. Phospholipid metabolism regulates the activity of these oncogenic signaling pathways to some extent. LPCAT1, a key enzyme involved in membrane phospholipid remodeling, when overexpressed, alters the lipid composition of the cell membrane, thereby promoting the activation of the MAPK/ERK signaling pathway, which enhances the stemness and metastatic potential of CSCs. Additionally, LPA and S1P activate YAP via G12/13-coupled receptors. S1P also activates the PI3K/AKT signaling pathway through binding to the G protein-coupled receptor S1PR. PA increases YAP activity by disrupting the formation of the LATS1-MOB1 complex and inhibiting the phosphorylation of LATS1/2. Inhibition of PA production via the PLD pathway can suppress the oncogenic activity of YAP
PI3K/AKT pathway is an integral part of glycerophospholipid metabolism and is often sustainably activated in CSC. PI3K can phosphorylate PIP2 to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3), both of which are phospholipid components of the cell membrane. As the second messenger, PIP3 promotes the recruitment of phosphoinositol-dependent kinase-1 (PDK1), leading to the activation of AKT. Once activated, the PI3K/AKT signal will be transmitted to a series of downstream pathways, the most classic of which is the mTOR pathway. mTOR is a key regulator of growth and metabolism. Under normal circumstances, mTOR inhibits autophagy, but under adverse circumstances such as stress, mTOR activation may promote cell survival—this can be a mechanism by which cancer cells adapt to stress [112–114]. In many cancers, the PI3K/AKT pathway is highly activated and involved in cancer cell proliferation, migration, EMT, and CSC phenotypes [115–118]. Recently, a study on osteosarcoma found that the PI3K/AKT pathway was associated with CSCs phenotype [119]. High expression of human epidermal growth factor receptor 2 (HER2) associated with colorectal cancer is also associated with aberrant activation of the PI3K/AKT pathway, and targeting PI3K/AKT can kill liver disseminated colorectal CSCs [120]. Cancer cells often contain a large amount of PIP3 due to the increased activity of oncogenic proteins upstream of PI3K or the activation of PI3K mutation [121]. It is worth noting that the phosphatase and tensin homolog deleted on chromosome ten (PTEN) gene is a tumor suppressor gene and a negative regulator of PI3K, which can block the downstream pathway regulated by AKT [122]. Some cancers also showed down-regulation and even loss of PTEN function [123]. These lead to the dysregulation of the PTEN/PI3K/AKT pathway, which is essential for maintaining prostate and breast CSCs [124,125]. In addition, S1P is an active sphingolipid metabolite, which can activate many tumor-related signal pathways, such as PI3K/AKT and RAS/ERK, by binding to the G protein-coupled S1P receptor (S1PR) [126].
The epidermal growth factor receptor (EGFR) is one of the most commonly amplified or mutated oncogenes in cancer. EGFR regulates cell proliferation and survival through downstream pathways, such as PI3K/AKT/mTOR and RAS/MAPK, and is related to stemness maintenance and drug resistance [127–129]. EGFR is a glycoprotein spanning the cell membrane and belongs to the tyrosine kinase receptor (TKR) family, including an extracellular ligand-binding region, a transmembrane (TM) domain, a juxtaposed membrane (JM) region containing tyrosine kinase, and C-terminal tail. The JM region plays a crucial role in the activation of EGFR. Research shows that PIP2 can regulate EGFR, and the JM region interacts with PIP2 [130]. The composition of membrane phospholipids can affect the EGFR signal by controlling the membrane structure, and the EGFR signal can also change the membrane properties. EGFR is overexpressed or abnormally expressed in many tumors. Dysregulated EGFR signals are essential in maintaining stem cell-like properties and driving tumor progression [131,132]. In EGFRvⅢ positive cancer cells, a significant decrease in lysophosphatidylcholine (LPC), and a notable increase in saturated PC, which helps sustain EGFR signaling and promote tumor growth [133]. This result is achieved through LPCAT1. A recent study found that silencing LPCAT1 can inhibit the growth of endometrial carcinoma. Overexpression of LPCAT1 changes phospholipid components such as PC and PE, leading to increased cancer cell stemness and metastasis [48]. In addition, LPCAT1 is necessary for the proliferation of many cancer cells, and it has become a potential therapeutic target [134]. Additionally, the EGFR pathway is overexpressed in drug-resistant breast stem cells [135]. In preclinical Inflammatory breast cancer (IBC) models, EGFR inhibitor treatment can reverse EMT and inhibit tumor growth and metastasis progression [136].
The Hippo pathway is evolutionarily conservative, including mammalian sterile 20-like kinase 1/2 (MST1/2), Salvador Family WW Domain Containing Protein 1 (SAV1), Large Tumor Suppressor Kinase 1/2 (LATS1/2), and Mps one Binder 1 (MOB1). MST1/2 phosphorylation activated LATS1/2, while LATS1/2 phosphorylation inhibited YAP and transcriptional coactivator with PDZ-binding motif (TAZ). YAP and TAZ are downstream effectors of the Hippo pathway, which are related to tumor cell proliferation, apoptosis, metastasis, and CSC properties [137]. The Hippo pathway restricts organ size by limiting cell growth and mediating stress-induced apoptosis, which are the keys to tumorigenesis. The Hippo/YAP pathway is aberrant in many solid tumors with enhanced YAP activity. For example, YAP is overexpressed in bladder cancer and is associated with cancer cell proliferation, migration, recurrence, and drug resistance [138]. The Hippo/YAP pathway is critical for maintaining the properties of CSCs, especially the downstream effector YAP. YAP is a crucial regulatory protein for CSC proliferation and carcinogenesis [139]. Studies have indicated that aberrant activation of YAP or TAZ maintains the survival, self-renewal, and multipotency of CSCs [140,141] and is highly associated with the EMT process. The Hippo pathway is abnormal in many cancers, and changes in the activity of its key components can lead to the occurrence and metastasis of tumors. It was found that LPA and S1P activated YAP through G 12/13 coupled receptors to regulate cell proliferation and tumorigenesis [142]. Han et al. found that PA also increases YAP activity by destroying the formation of the LATS1-MOB1 complex and inhibiting LATS1/2 phosphorylation. They also found that inhibition of PA production, specifically through the PLD pathway, can suppress the carcinogenic activity of YAP so that the PLD-PA-YAP axis may become a potential therapeutic target [143]. Phospholipase C Delta3 (PLCD3) is an oncogene related to the occurrence and progression of thyroid cancer and is related to the Hippo pathway. When PLCD3 was downregulated, the expression of YAP1/TAZ decreased significantly. PLCD3 silencing can also reverse EMT and induce apoptosis, but overexpression of YAP1 can compensate for this tumor inhibition [144]. Moreover, the Hippo pathway can also be regulated by sphingomyelin phosphate choline in a dual and reverse manner [145]. SCD1 has also been linked to YAP/TAZ expression in CSCs, adding another layer to the complex interplay between phospholipid metabolism and Hippo pathway signaling in cancer [146].
Ferroptosis is a new type of death driven by intracellular iron accumulation, membrane phospholipid peroxidation, and increased ROS. Many studies have shown that ferroptosis is related to various diseases, including cancers and ischemic diseases [147–149]. Ferroptosis is also closely related to the phenotype of CSCs. For instance, CD44, a surface marker of CSC, is closely associated with the resistance of CSCs to ferroptosis. For example, the expression of variant subtype CD44v helps CSCs defend against ROS by promoting the synthesis of reduced glutathione (GSH) [150]. Similarly, studies have found that the overexpression of CD44 enhances the stability of Solute carrier family 7 member 11 (SLC7A11), a key component of the cystine/glutamate antiporter, and inhibits ferroptosis by promoting the interaction between SLC7A11 and ovarian tumor associated proteinase B1 (OTUB1), an ovarian tumor family member deubiquitinase [151]. The substrate of lipid peroxidation is PUFAs. Studies have shown that PE containing PUFA is the essential phospholipid for lipid peroxidation and ferroptosis [152]. The research indicates that LPCAT3 inhibitors reshape the content of membrane polyunsaturated phospholipids and partially prevent cell ferroptosis [109]. Simultaneously, some MUFAs also produce similar effects [30]. Furthermore, CSCs can actively regulate their differentiation status to avoid the adverse effects of ferroptosis. For example, YAP/TAZ is involved in cancer cells avoiding ferroptosis. The metabolism and utilization of iron in CSCs were significantly accelerated, and the iron concentration in CSCs was higher than in non-CSCs. When irons in cells increase, phospholipids containing PUFA are more likely to be peroxidized, resulting in the production of excessive ROS [153]. High levels of ROS can cause the loss of self-renewal and pluripotency of CSCs and enhance their radiosensitivity [154,155]. The intervention of ROS-regulating drugs on drug-resistant CSCs may have better therapeutic effects. Inhibition of ferritin expression can also impair the growth of GSCs [156]. In addition, many glutathione peroxidases (GPX) function in the proliferation, drug resistance, metastasis, and recurrence of CSCs. GSH is converted into oxidized glutathione (GSSG) by GPX4 and is used to consume intracellular peroxides. Therefore, inhibition of System Xc− and GPX4 will accumulate lipid peroxides, leading to ferroptosis [157]. In addition, mesenchymal CSC populations associated with mesenchymal tumors are susceptible to ferroptosis [158].
CSCs are a subset of cancer cells in the state of stem cells with stem cell characteristics. Studies have shown that CSCs widely exist in different types of tumors. Most tumor cells can be removed by radiotherapy and chemotherapy, but the critical oncogenic CSCs survive, enabling them to induce tumor recurrence and metastasis. The ultimate result of CSC research is to identify the pathways that selectively regulate CSCs and eradicate CSCs for these pathways. Studies have shown that the phospholipid metabolism pattern of CSCs is distinct from the others. Non-cancerous stem cells, such as those found in tissues like the bone marrow or brain, exhibit relatively stable and balanced metabolic profiles. Their phospholipid metabolism primarily supports the maintenance of cell pluripotency, differentiation potential, and tissue homeostasis. Compared to cancer cells, the overall metabolism of these stem cells is less dynamic. These stem cells primarily produce phospholipids required for stem cell maintenance and differentiation, rather than actively growing or surviving in hostile microenvironments. In contrast, CSCs often exhibit altered phospholipid metabolism as part of tumor heterogeneity. CSCs modulate their phospholipid metabolism to support rapid proliferation, survival, and resistance to therapy. To adapt to the tumor microenvironment, which may involve hypoxia, nutrient deprivation, and immune challenges, the metabolism of CSCs becomes more dynamic. This is mainly manifested in the upregulation of phospholipid synthesis, desaturation, and FAO. Therefore, the phospholipid metabolism of CSCs not only differs significantly from that of normal stem cells, but also provides key survival mechanisms that support their persistence, proliferation, and resistance to conventional cancer therapies. These unique metabolic characteristics make CSCs a promising target for the development of new and more effective cancer treatments. Therefore, a comprehensive and clear understanding of the biological characteristics and stem maintenance mechanism of CSCs is necessary to develop tumor therapy. This review introduces the types of CSCs and some surface markers, summarizes the unique phospholipid metabolism reprogramming of CSCs, and discusses the main ways to regulate the stemness characteristics of CSCs.
The rapid proliferation of tumor cells leads to excessive demand for energy supply and structural components, and significant changes have occurred in cellular metabolism. Phospholipid metabolic reprogramming is closely related to the characteristics of CSCs, which is mainly manifested in the upregulation of phospholipid synthesis, desaturation, and lipolysis-FAO. Targeting lipid metabolism has become a feasible strategy for therapeutic intervention. In addition, the combination of phospholipid metabolism-related inhibitors with chemotherapy drugs or immunotherapy can improve the anti-cancer effect. FASN is a major focus in targeting CSCs, and the development of novel FASN inhibitors is underway. The antitumor activity of FASN inhibitors has been confirmed in preclinical cancer models. For example, the novel FASN inhibitor AZ12756122, which targets FASN, represents a potential treatment option to overcome resistance to EGFR tyrosine kinase inhibitors [159]. Preclinical studies showed that combined with Osimertinib, it could synergistically reduce characteristics of the CSC in EGFR mutant NSCLC cells. This further highlights the importance and potential of FASN inhibitors in inhibiting CSCs and combating cancer. In addition, selective elimination of CSCs can be achieved by targeting SCD1. One study suggests that targeting SCD1 selectively eliminates colon CSCs by inhibiting Wnt and NOTCH signaling pathways [160]. SCD inhibitors, including A939572 and the novel compounds CVT-11127 or CVT-12012, have been studied for their effects on membrane lipid saturation and signal transduction, providing a new approach to fight against the chemotherapy resistance of cancer cells [161]. Additionally, the dependence of CSCs on FAO further justifies the rationale of targeting these cells with FAO inhibitors. Compounds like Etomoxir, which inhibit FAO, have shown potential in preclinical studies for slowing tumor growth, particularly in gliomas [162]. Furthermore, inhibition of PLD can suppress the carcinogenic activity of YAP, thereby regulating EMT and stemness. Therefore, PLD inhibitors are also a research hotspot. PLC is abnormally expressed in GSCs and LSCs, which may provide an opportunity to target specific signaling nodes for CSCs in leukemia and glioma. The research shows that combining clinical therapeutic drugs, such as cisplatin, with PLA2 inhibitors is expected to eliminate BCSCs and tumorigenesis effectively [163]. In the animal experimental model by Singh et al., using Arf1 inhibitors can reduce the formation of CSCs, and the impact on normal stem cells can be ignored [65]. Wang et al. also found that knocking down ARF1 could not only kill CSCs but also trigger tumor-specific immune responses. The dying CSCs were transformed into therapeutic vaccines to attract and activate immune cells to destroy large tumors and lead to lasting therapeutic effects [164]. These findings suggest that targeting lipid metabolism could provide a valuable therapeutic strategy for overcoming resistance and controlling tumor progression. Another strategy some researchers propose is building engineered fat cells to deliver anti-cancer drugs [165,166]. This approach allows for selective targeting and efficient elimination, enhancing the precision and effectiveness of cancer treatment.
Inhibition of the PI3K/AKT pathway has always been a research focus of targeted therapy. Tumors with abnormal PI3K/AKT pathway mutations may benefit from PI3K/AKT inhibitor-mediated molecular targeted therapy. Many small molecule inhibitors of the PI3K/AKT/mTOR signaling pathway have been studied in the preclinical stage. However, only a few PI3K and mTOR inhibitors have been approved for the treatment of human cancer in the clinic. Monotherapy is gradually changing to combination therapy, which has become a significant research direction [167]. At present, the regulatory role of S1P and DAG in the PI3K/AKT pathway may become potential research targets for cancer therapy. In recent years, new advances have been made in KRAS-targeted drugs, especially in KRAS-G12C inhibitors, such as AMG510 (Sotorasib) and MRTX849 (Adagrasib), both of which have been approved for clinical use [168]. Several RAF inhibitors have also demonstrated superior clinical efficacy in patients with RAF mutant cancers. The Hippo pathway is regulated by dual and reverse regulation of LPA, S1P, and phosphatidylcholine phosphate. Abnormal expression of SCD1 is also associated with YAP/TAZ in CSC, highlighting further potential targets for therapeutic intervention. Studies have found that the anti-tumor effect can be enhanced by inducing ferroptosis of tumor cells, and this anti-cancer strategy has broad prospects [169,170]. Disrupting the redox balance of CSCs through targeted use of ROS-regulating drugs will become the latest direction of cancer treatment research. Numerous studies have explored drugs and drug delivery systems designed to provide precise platforms for selectively targeting and killing tumor cells, offering significant potential for improving cancer treatment outcomes [171,172].
Phospholipid reprogramming is a key metabolic change that supports the rapid growth and survival of tumor cells. This process is closely related to the Warburg effect and glycolysis, both of which contribute to the altered metabolic patterns of cancer cells. Phospholipid reprogramming involves several key alterations: increased phospholipid synthesis, enhanced desaturation of phospholipids, and increased FA uptake and FAO. The Warburg effect is characterized by cancer cells preferentially using glycolysis to generate ATP, a shift that has significant implications for phospholipid metabolism. On one hand, the glycolytic pathway provides essential intermediates for phospholipid biosynthesis. For example, glycerol-3-phosphate is used to synthesize glycerophospholipids, including PC. Therefore, the upregulation of glycolysis in cancer cells directly contributes to increased phospholipid synthesis. On the other hand, the Warburg effect, by shifting cellular metabolism to glycolysis, also promotes the production of metabolic precursors. Acetyl-CoA, a precursor for fatty acid and phospholipid biosynthesis, is one such example. In cancer, glycolysis is often upregulated, leading to an increased production of acetyl-CoA, which further drives phospholipid synthesis.
In addition, there may be other compensation pathways in tumor tissues that can protect CSCs from energy deficiency and binding of signal intermediates. Therefore, we need more research on the heterogeneity of phospholipid metabolism in CSCs. In general, due to these metabolic changes’ relative measurability and operability, some progress has been made in targeting phospholipid metabolism, inhibiting CSCs, reducing drug resistance, and other aspects in recent years. Further research on its regulatory mechanism is expected to develop targeted drugs to minimize drug resistance or new therapeutic strategies and synergistic combination therapies. An important concern related to this issue is the problem of cell models in in vitro studies, especially when it involves metabolism-related mechanisms. In the tumor microenvironment, factors such as hypoxia, abnormal vasculature, and competition among various cell types for limited nutrients contribute to the heterogeneity of tumor cells and are also factors that promote and maintain stemness in tumor cells. However, in in vitro studies, even when using nutrient-restricted media, there are still significant differences compared to the environment in which tumor cells exist in vivo. Even with the increasingly used organoid models, our experience shows that the tumor cells capable of forming tumor organoids are usually those with higher stemness, and the proportion of such cells is very high, which also differs from in vivo observations. Therefore, exploring new in vitro stem cell research models is a very important aspect of future research.
CSCs are a subgroup of cancer cells that have the characteristics of stem cells. CSCs are prevalent in various tumor types and can relapse and metastasize, unlike other tumor cells that are usually eliminated by radiation and chemotherapy. CSCs have a unique pattern of phospholipid metabolism that differs from that of other cancer cells: increased phospholipid synthesis, phospholipid desaturation, and lipolysis-FAO. Whether it is targeting phospholipid metabolites or related metabolic enzymes and the stemness signal axis of activation regulation, it opens the possibility for the development of targeted therapies aimed at these cells.
Acknowledgement: None.
Funding Statement: This work was supported by grants from National Natural Science Foundation of China (81902784, 81771086), by the Fund of Sichuan Provincial Department of science and technology (2024YFFK0393, 2022YFS0039), the Research and Develop Program, West China Hospital of Stomatology, Sichuan University (LCYJ2023-DL-2, RD-02-202002), the CAMS Innovation Fund for Medical Sciences (CIFMS, 2019-I2M-5-004). The funding agencies had no role in the study design, collection, analysis, or interpretation of data, writing of the report, or the decision to submit the article for publication.
Author Contributions: The authors confirm their contribution to the paper as follows: draft manuscript preparation: Qing Wang; review and editing: Yu Zhou, Yingqiang Shen; visualization: Luyao Cai, Shouyi Tang, Dan Pan, Zhen Wang; supervision: Qianming Chen, Yu Zhou, Yingqiang Shen. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
Abbreviations
| ACC | Acetyl CoA carboxylase |
| ACLY | ATP citrate lyase |
| ACS | Acyl CoA synthetase |
| ACSLS | Long-chain acyl CoA synthetases |
| AKT | Protein kinase B |
| ALDH1 | Aldehyde dehydrogenase 1 |
| ASM | Acid sphingomyelinase |
| BCAP | B-cell adaptor protein |
| BCR | B cell receptor |
| BCSCs | Brain cancer stem cells |
| CACT | Carnitine acylcarnitine translocase |
| CDP | Cytidine diphosphate |
| CHKα | Choline kinase α |
| CHOP | C/EBP homologous protein |
| CL | Cardiolipin |
| COPI | Coat protein complex I |
| CPT1 | Carnitine palmitoyltransferase 1 |
| CSCs | Cancer stem cells |
| DAG | Diglyceride |
| DHAP | Dihydroxyacetone phosphate |
| EGFR | Epidermal growth factor receptor |
| ELOVL | Extended by elongases of very long-chain fatty acids |
| EMT | Epithelial-mesenchymal transformation |
| EOCs | Epithelial ovarian cancers |
| ERK | Extracellular regulated protein kinases |
| ETNK | Ethanolamine kinase |
| FA | Fatty acid |
| FABP | Fatty acid binding protein |
| FADS2 | Fatty acid desaturase 2 |
| FAO | Fatty acid oxidation |
| FASN | Fatty acid synthase |
| FGFR | Fibroblast growth factor receptor |
| FRS2 | FGF-receptor substrate 2 |
| GAB1 | GRB2-associated binder 1 |
| GCSCs | Gastric cancer stem cells |
| GPCR | G Protein-Coupled Receptors |
| GPX | Glutathione peroxidases |
| GRB2 | Growth factor receptor-bound protein 2 |
| GSCs | Glioma stem cells |
| GSH | Reduced glutathione |
| GSSG | Oxidized glutathione |
| HER2 | Human epidermal growth factor receptor 2 |
| HIF-1 | Hypoxia-inducible factor 1 |
| IBC | Inflammatory breast cancer |
| IFN | Interferon |
| JM | Juxtaposed membrane |
| LATS1/2 | Large Tumor Suppressor Kinase 1/2 |
| LCFAs | Long-chain FAs |
| LncRNA | Long non-coding RNA |
| LPA | Lysophosphatidic acid |
| LPC | Lysophosphatidylcholine |
| LPCAT | Lysophosphatidylcholine acyltransferase |
| LSCs | Leukemia stem cells |
| MAPK | Mitogen-activated protein kinase |
| MARK4 | Microtubule affinity regulating kinase 4 |
| MEK | Mitogen-activated protein kinase kinase |
| MOB1 | Mps one Binder 1 |
| mTOR | Mammalian target of Rapamycin |
| MUFAs | Monounsaturated FAs |
| NSCLC | Non-small cell lung cancer |
| nSMase | Neutral sphingomyelinase |
| OTUB1 | Ovarian tumor associated proteinase B1 |
| PA | Phosphatidic acid |
| PaCSCs | Pancreatic cancer stem cells |
| PAP | Phosphatidic acid phosphatase |
| PC | Phosphatidylcholine |
| PDK1 | Phosphoinositol-dependent kinase-1 |
| PE | Phosphatidylethanolamine |
| PI | Phosphatidylinositol |
| PI3K | Phosphatidylinositol-3-kinase |
| PIP2 | Phosphatidylinositol-45-bisphosphate |
| PIP3 | Phosphatidylinositol 3,4,5-trisphosphate |
| PLA | Phospholipase A |
| PLC | Phospholipase C |
| PLCD3 | Phospholipase C Delta3 |
| PLCγ1 | Phospholipase C Gamma 1 |
| PLD | Phospholipase D |
| PS | Phosphatidylserine |
| PTEN | Phosphatase and tensin homolog deleted on chromosome ten |
| PUFAs | Polyunsaturated FAs |
| Rheb | Ras homolog enriched in brain |
| ROS | Reactive oxygen species |
| RSK | Ribosomal S6 Kinase |
| S1P | Sphingosine 1 phosphate |
| S1PR | S1P receptor |
| SASP | Senescence-associated secretory phenotype |
| SAV1 | Salvador Family WW Domain Containing Protein 1 |
| SCD | Stearoyl COA desaturase |
| SHC | SH2-containing sequence |
| SK1 | Sphingosine kinase 1 |
| SLC7A11 | Solute carrier family 7 member 11 |
| SM | Sphingomyelinase |
| SOS | Son of Sevenless |
| SOX2 | SRY-box transcription factor 2 |
| SPHK | Sphingosine kinase |
| sPLA2 | Secreted phospholipase A2 |
| SRS | Stimulated Raman scattering |
| STAT1 | Signal transducer and activator of transcription 1 |
| TAZ | Transcriptional coactivator with PDZ-binding motif |
| TCA | Tricarboxylic acid |
| TCR | T cell receptor |
| TKR | Tyrosine kinase receptor |
| TM | Transmembrane |
| TNBC | Triple-negative breast cancer |
| TSC1/2 | Tuberous sclerosis complex 1/2 |
| UFAs | Unsaturated FAs |
| YAP | Yes-associated protein |
References
1. Han X, Du F, Jiang L, Zhu Y, Chen Z, Liu Y, et al. [Corrigendum] A2780 human ovarian cancer cells with acquired paclitaxel resistance display cancer stem cell properties. Oncol Lett. 2022;24(5):406. doi:10.3892/ol.2022.13526. [Google Scholar] [PubMed] [CrossRef]
2. Zhang R, Zhang X, Zhang W, Cui W, Xiao Y, Liu L et al. Sohlh2 regulates the stemness and differentiation of colon cancer stem cells by downregulating LncRNA-H19 transcription. Mol Cancer Res. 2022;21(2):115–26. doi:10.1158/1541-7786.MCR-22-0134. [Google Scholar] [PubMed] [CrossRef]
3. Rajabi A, Kayedi M, Rahimi S, Dashti F, Mirazimi SMA, Homayoonfal M, et al. Non-coding RNAs and glioma: focus on cancer stem cells. Mol Ther-Oncolytics. 2022;27:100–23. doi:10.1016/j.omto.2022.09.005. [Google Scholar] [PubMed] [CrossRef]
4. Yin J, Wang Y, Wei G, Wen M. UBE2T mediates the stemness properties of breast cancer cells through the mTOR signaling pathway. BIOCELL. 2024;48:959–70. doi:10.32604/biocell.2024.049349. [Google Scholar] [CrossRef]
5. Yang D, Dai Z, Zhu P, Wang G, Sun B, Li S, et al. High throughput-screening of native herbal compounds identifies taccaoside A as a cytotoxic compound that mediates RAS signaling in cancer stem cells. Phytomedicine. 2023;108:154492. doi:10.1016/j.phymed.2022.154492. [Google Scholar] [PubMed] [CrossRef]
6. Xu Z. CD133+ cancer stem cells in lung cancer. Front Biosci. 2013;18(2):447–53. doi:10.2741/4113. [Google Scholar] [PubMed] [CrossRef]
7. Rabinovich I, Sebastião APM, Lima RS, Urban CDA, Junior ES, Anselmi KF et al. Cancer stem cell markers ALDH1 and CD44+/CD24– phenotype and their prognosis impact in invasive ductal carcinoma. Eur J Histochem EJH. 2018;62(3):2943. doi:10.4081/ejh.2018.2943. [Google Scholar] [PubMed] [CrossRef]
8. Zeng C, Zhang Y, Park SC, Eun JR, Nguyen NT, Tschudy-Seney B, et al. CD34+ liver cancer stem cells were formed by fusion of hepatobiliary stem/progenitor cells with hematopoietic precursor-derived myeloid intermediates. Stem Cells Dev. 2015;24(21):2467–78. doi:10.1089/scd.2015.0202. [Google Scholar] [PubMed] [CrossRef]
9. Luo W, Zhang D, Ma S, Wang C, Zhang Q, Wang H et al. miR-27a is highly expressed in H1650 cancer stem cells and regulates proliferation, migration, and invasion. J Cancer Res Ther. 2018;14:S1004–11. doi:10.4103/0973-1482.199450. [Google Scholar] [PubMed] [CrossRef]
10. Pustovalova M, Blokhina T, Alhaddad L, Chigasova A, Chuprov-Netochin R, Veviorskiy A, et al. CD44+ and CD133+ non-small cell lung cancer cells exhibit DNA damage response pathways and dormant polyploid giant cancer cell enrichment relating to their p53 status. Int J Mol Sci. 2022;23(9):4922. doi:10.3390/ijms23094922. [Google Scholar] [PubMed] [CrossRef]
11. Cioffi M, D’Alterio C, Camerlingo R, Tirino V, Consales C, Riccio A, et al. Identification of a distinct population of CD133+CXCR4+ cancer stem cells in ovarian cancer. Sci Rep. 2015;5:10357. doi:10.1038/srep10357. [Google Scholar] [PubMed] [CrossRef]
12. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946–51. doi:10.1158/0008-5472.CAN-05-2018. [Google Scholar] [PubMed] [CrossRef]
13. Sun JH, Luo Q, Liu LL, Song GB. Liver cancer stem cell markers: progression and therapeutic implications. World J Gastroenterol. 2016;22(13):3547–57. doi:10.3748/wjg.v22.i13.3547. [Google Scholar] [PubMed] [CrossRef]
14. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi:10.1038/nature03128. [Google Scholar] [PubMed] [CrossRef]
15. de Beça FF, Caetano P, Gerhard R, Alvarenga CA, Gomes M, Paredes J et al. Cancer stem cells markers CD44, CD24 and ALDH1 in breast cancer special histological types. J Clin Pathol. 2013;66:187–91. doi:10.1136/jclinpath-2012-201169. [Google Scholar] [PubMed] [CrossRef]
16. Skoda J, Hermanova M, Loja T, Nemec P, Neradil J, Karasek P, et al. Co-expression of cancer stem cell markers corresponds to a pro-tumorigenic expression profile in pancreatic adenocarcinoma. PLoS One. 2016;11:e0159255. doi:10.1371/journal.pone.0159255. [Google Scholar] [PubMed] [CrossRef]
17. Gómez-Gallegos AA, Ramírez-Vidal L, Becerril-Rico J, Pérez-Islas E, Hernandez-Peralta ZJ, Toledo-Guzmán ME, et al. CD24+CD44+CD54+EpCAM+ gastric cancer stem cells predict tumor progression and metastasis: clinical and experimental evidence. Stem Cell Res Ther. 2023;14(1):16. doi:10.1186/s13287-023-03241-7. [Google Scholar] [PubMed] [CrossRef]
18. Lee TKW, Castilho A, Cheung VCH, Tang KH, Ma S, Ng IOL. CD24+ liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011;9:50–63. doi:10.1016/j.stem.2011.06.005. [Google Scholar] [PubMed] [CrossRef]
19. El-Ashmawy NE, Salem ML, Abd El-Fattah EE, Khedr EG. Targeting CD166 lung cancer stem cells: molecular study using murine dendritic cell vaccine+. Toxicol Appl Pharmacol. 2021;429:115699. doi:10.1016/j.taap.2021.115699. [Google Scholar] [PubMed] [CrossRef]
20. Kalantari E, Taheri T, Fata S, Abolhasani M, Mehrazma M, Madjd Z, et al. Significant co-expression of putative cancer stem cell markers, EpCAM and CD166, correlates with tumor stage and invasive behavior in colorectal cancer. World J Surg Oncol. 2022;20:15. doi:10.1186/s12957-021-02469-y. [Google Scholar] [PubMed] [CrossRef]
21. Nguyen PH, Giraud J, Chambonnier L, Dubus P, Wittkop L, Belleannée G, et al. Characterization of biomarkers of tumorigenic and chemoresistant cancer stem cells in human gastric carcinoma. Clin Cancer Res off J Am Assoc Cancer Res. 2017;23:1586–97. doi:10.1158/1078-0432.CCR-15-2157. [Google Scholar] [PubMed] [CrossRef]
22. Bohdanowicz M, Grinstein S. Role of phospholipids in endocytosis, phagocytosis, and macropinocytosis. Physiol Rev. 2013;93:69–106. doi:10.1152/physrev.00002.2012. [Google Scholar] [PubMed] [CrossRef]
23. Yang Y, Xu J, Su Q, Wu Y, Li Q, Ma Z, et al. Lysophosphatidic acid induced apoptosis, DNA damage, and oxidative stress in spinal cord neurons by upregulating LPA4/LPA6 receptors. Mediators Inflamm. 2022;2022:1–15. doi:10.1155/2022/1818758. [Google Scholar] [PubMed] [CrossRef]
24. Ogawa T, Kuboshima M, Suwanawat N, Kawamoto J, Kurihara T. Division of the role and physiological impact of multiple lysophosphatidic acid acyltransferase paralogs. BMC Microbiol. 2022;22:241. doi:10.1186/s12866-022-02641-8. [Google Scholar] [PubMed] [CrossRef]
25. Li H, Feng Z, He ML. Lipid metabolism alteration contributes to and maintains the properties of cancer stem cells. Theranostics. 2020;10(16):7053–69. doi:10.7150/thno.41388. [Google Scholar] [PubMed] [CrossRef]
26. Yi M, Li J, Chen S, Cai J, Ban Y, Peng Q, et al. Correction to: emerging role of lipid metabolism alterations in cancer stem cells. J Exp Clin Cancer Res. 2018;37(1):155. doi:10.1186/s13046-018-0826-z. [Google Scholar] [PubMed] [CrossRef]
27. Di Carlo C, Sousa BC, Manfredi M, Brandi J, Dalla Pozza E, Marengo E, et al. Integrated lipidomics and proteomics reveal cardiolipin alterations, upregulation of HADHA and long chain fatty acids in pancreatic cancer stem cells. Sci Rep. 2021;11:13297. doi:10.1038/s41598-021-92752-5. [Google Scholar] [PubMed] [CrossRef]
28. Guan Y, Chen X, Wu M, Zhu W, Arslan A, Takeda S, et al. The phosphatidylethanolamine biosynthesis pathway provides a new target for cancer chemotherapy. J Hepatol. 2020;72:746–60. doi:10.1016/j.jhep.2019.11.007. [Google Scholar] [PubMed] [CrossRef]
29. Bian X, Liu R, Meng Y, Xing D, Xu D, Lu Z. Lipid metabolism and cancer. J Exp Med. 2021;218:e20201606. doi:10.1084/jem.20201606. [Google Scholar] [PubMed] [CrossRef]
30. Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. 2020;31:62–76. doi:10.1016/j.cmet.2019.11.010. [Google Scholar] [PubMed] [CrossRef]
31. Nakagawa H, Hayata Y, Kawamura S, Yamada T, Fujiwara N, Koike K. Lipid metabolic reprogramming in hepatocellular carcinoma. Cancers. 2018;10:447. doi:10.3390/cancers10110447. [Google Scholar] [PubMed] [CrossRef]
32. Chen Y, Li P. Fatty acid metabolism and cancer development. Sci Bull. 2016;61(19):1473–9. doi:10.1007/s11434-016-1129-4. [Google Scholar] [CrossRef]
33. Currie E, Schulze A, Zechner R, Walther TC, Farese RV. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153–61. doi:10.1016/j.cmet.2013.05.017. [Google Scholar] [PubMed] [CrossRef]
34. Blassberg R, Jacob J. Lipid metabolism fattens up hedgehog signaling. BMC Biol. 2017;15(1):95. doi:10.1186/s12915-017-0442-y. [Google Scholar] [PubMed] [CrossRef]
35. Wang B, Tontonoz P. Phospholipid remodeling in physiology and disease. Annu Rev Physiol. 2019;81:165–88. doi:10.1146/annurev-physiol-020518-114444. [Google Scholar] [PubMed] [CrossRef]
36. Kato D, Aoyama Y, Nishida K, Takahashi Y, Sakamoto T, Takeda I, et al. Regulation of lipid synthesis in myelin modulates neural activity and is required for motor learning. Glia. 2023;71:2591–608. doi:10.1002/glia.24441. [Google Scholar] [PubMed] [CrossRef]
37. Viale A, Pettazzoni P, Lyssiotis CA, Ying HQ, Sánchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514(7524):628–32. doi:10.1038/nature13611. [Google Scholar] [PubMed] [CrossRef]
38. Lue HW, Podolak J, Kolahi K, Cheng L, Rao S, Garg D, et al. Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes Dev. 2017;31(20):2067–84. doi:10.1101/gad.305292.117. [Google Scholar] [PubMed] [CrossRef]
39. Di Francesco AM, Toesca A, Cenciarelli C, Giordano A, Gasbarrini A, Puglisi MA. Metabolic modification in gastrointestinal cancer stem cells: characteristics and therapeutic approaches. J Cell Physiol. 2016;231(10):2081–7. doi:10.1002/jcp.25318. [Google Scholar] [PubMed] [CrossRef]
40. He J, Zhang F, Rachel Tay LW, Boroda S, Nian W, Levental KR, et al. Lipin-1 regulation of phospholipid synthesis maintains endoplasmic reticulum homeostasis and is critical for triple-negative breast cancer cell survival. FASEB J. 2017;31:2893–904. doi:10.1096/fj.201601353R. [Google Scholar] [PubMed] [CrossRef]
41. Mohan A, Raj Rajan R, Mohan G, Kollenchery Puthenveettil P, Maliekal TT. Markers and reporters to reveal the hierarchy in heterogeneous cancer stem cells. Front Cell Dev Biol. 2021;9:668851. doi:10.3389/fcell.2021.668851. [Google Scholar] [PubMed] [CrossRef]
42. Kubota CS, Espenshade PJ. Targeting Stearoyl-CoA desaturase in solid tumors. Cancer Res. 2022;82(9):1682–8. doi:10.1158/0008-5472.CAN-21-4044. [Google Scholar] [PubMed] [CrossRef]
43. Raggi C, Taddei ML, Rae C, Braconi C, Marra F. Metabolic reprogramming incholangiocarcinoma. J Hepatol. 2022;77(3):849–64. doi:10.1016/j.jhep.2022.04.038. [Google Scholar] [PubMed] [CrossRef]
44. Rabionet M, Polonio-Alcalá E, Relat J, Yeste M, Sims-Mourtada J, Kloxin AM, et al. Fatty acid synthase as a feasible biomarker for triple negative breast cancer stem cell subpopulation cultured on electrospun scaffolds. Mater Today Bio. 2021;12:100155. doi:10.1016/j.mtbio.2021.100155. [Google Scholar] [PubMed] [CrossRef]
45. Liu HH, Xu Y, Li CJ et al. An SCD1-dependent mechanoresponsive pathway promotes HCC invasion and metastasis through lipid metabolic reprogramming. Mol Ther. 2022;30(7):2554–67. doi:10.1016/j.ymthe.2022.03.015. [Google Scholar] [PubMed] [CrossRef]
46. Seo EJ, Kwon YW, Jang IH, Kim DK, Lee SI, Choi EJ, et al. Autotaxin regulates maintenance of ovarian cancer stem cells through lysophosphatidic acid-mediated autocrine mechanism. Stem Cells. 2016;34:551–64. doi:10.1002/stem.2279. [Google Scholar] [PubMed] [CrossRef]
47. Wang B, Rong X, Palladino END, Wang J, Fogelman AM, Martín MG, et al. Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell. 2018;22:206–20. doi:10.1016/j.stem.2017.12.017. [Google Scholar] [PubMed] [CrossRef]
48. Zhao T, Sun R, Ma X, Wei L, Hou Y, Song K, et al. Overexpression of LPCAT1 enhances endometrial cancer stemness and metastasis by changing lipid components and activating the TGF/β-Smad2/3 signaling pathway. Acta Biochim Biophys Sin. 2022;54:904–16. doi:10.3724/abbs.2022076. [Google Scholar] [PubMed] [CrossRef]
49. Song M, Lee H, Nam M-H, Jeong E, Kim S, Hong Y, et al. Loss-of-function screens of druggable targetome against cancer stem-like cells. FASEB J off Publ Fed Am Soc Exp Biol. 2017;31:625–35. doi:10.1096/fj.201600953. [Google Scholar] [PubMed] [CrossRef]
50. Sun M, Yang Z. Metabolomic studies of live single cancer stem cells using mass spectrometry. Anal Chem. 2019;91:2384–91. doi:10.1021/acs.analchem.8b05166. [Google Scholar] [PubMed] [CrossRef]
51. Gimple RC, Kidwell RL, Kim LJY, Sun T, Gromovsky AD, Wu Q, et al. Glioma stem cell-specific superenhancer promotes polyunsaturated fatty-acid synthesis to support EGFR signaling. Cancer Discov. 2019;9:1248–67. doi:10.1158/2159-8290.CD-19-0061. [Google Scholar] [PubMed] [CrossRef]
52. J L, S C, J T-P, X M, Y X, Td H et al. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell. 2017;20:303–14.e5. doi:10.1016/j.stem.2016.11.004. [Google Scholar] [PubMed] [CrossRef]
53. Xuan Y, Wang H, Yung MM, Chen F, Chan WS, Chan YS, et al. SCD1/FADS2 fatty acid desaturases equipoise lipid metabolic activity and redox-driven ferroptosis in ascites-derived ovarian cancer cells. Theranostics. 2022;12(7):3534–52. doi:10.7150/thno.70194. [Google Scholar] [PubMed] [CrossRef]
54. Colacino JA, McDermott SP, Sartor MA, Wicha MS, Rozek LS. Transcriptomic profiling of curcumin-treated human breast stem cells identifies a role for stearoyl-coa desaturase in breast cancer prevention. Breast Cancer Res Treat. 2016;158:29–41. doi:10.1007/s10549-016-3854-4. [Google Scholar] [PubMed] [CrossRef]
55. Gao Y, Li J, Xi H, Cui J, Zhang K, Zhang J, et al. Stearoyl-CoA-desaturase-1 regulates gastric cancer stem-like properties and promotes tumour metastasis via Hippo/YAP pathway. Br J Cancer. 2020;122:1837–47. doi:10.1038/s41416-020-0827-5. [Google Scholar] [PubMed] [CrossRef]
56. Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A, et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature. 2019;566(7744):403–6. doi:10.1038/s41586-019-0904-1. [Google Scholar] [PubMed] [CrossRef]
57. Guth A, Monk E, Agarwal R, Bergman BC, Zemski-Berry KA, Minic A, et al. Targeting fat oxidation in mouse prostate cancer decreases tumor growth and stimulates anti-cancer immunity. Int J Mol Sci. 2020;21:E9660. doi:10.3390/ijms21249660. [Google Scholar] [PubMed] [CrossRef]
58. Lu R, Hong J, Fu T, Zhu Y, Tong R, Ai D, et al. Loss of OVOL2 in triple-negative breast cancer promotes fatty acid oxidation fueling stemness characteristics. Adv Sci. 2024;11:2308945. doi:10.1002/advs.202308945. [Google Scholar] [PubMed] [CrossRef]
59. Wang T, Fahrmann JF, Lee H, Li Y-J, Tripathi SC, Yue C, et al. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018;27:136–50. doi:10.1016/j.cmet.2017.11.001. [Google Scholar] [PubMed] [CrossRef]
60. Chen C-L, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, et al. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab. 2016;23:206–19. doi:10.1016/j.cmet.2015.12.004. [Google Scholar] [PubMed] [CrossRef]
61. Yu L, Wei W, Lv J, Lu Y, Wang Z, Cai C. FABP4-mediated lipid metabolism promotes TNBC progression and breast cancer stem cell activity. Cancer Lett. 2024;604:217271. doi:10.1016/j.canlet.2024.217271. [Google Scholar] [PubMed] [CrossRef]
62. Mascaraque M, Courtois S, Royo-García A, Barneda D, Stoian AM, Villaoslada I, et al. Fatty acid oxidation is critical for the tumorigenic potential and chemoresistance of pancreatic cancer stem cells. J Transl Med. 2024;22:797. doi:10.1186/s12967-024-05598-6. [Google Scholar] [PubMed] [CrossRef]
63. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. 2016;19:23–37. doi:10.1016/j.stem.2016.06.001. [Google Scholar] [PubMed] [CrossRef]
64. Hale JS, Otvos B, Sinyuk M, Alvarado AG, Hitomi M, Stoltz K, et al. Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells Dayt Ohio. 2014;32:1746–58. doi:10.1002/stem.1716. [Google Scholar] [PubMed] [CrossRef]
65. Singh SR, Zeng X, Zhao J, Liu Y, Hou G, Liu H, et al. The lipolysis pathway sustains normal and transformed stem cells in adult Drosophila. Nature. 2016;538:109–13. doi:10.1038/nature19788. [Google Scholar] [PubMed] [CrossRef]
66. Mahalingaiah PKS, Ponnusamy L, Singh KP. Chronic oxidative stress leads to malignant transformation along with acquisition of stem cell characteristics, and epithelial to mesenchymal transition in human renal epithelial cells. J Cell Physiol. 2015;230:1916–28. doi:10.1002/jcp.24922. [Google Scholar] [PubMed] [CrossRef]
67. Cheng M, Bhujwalla ZM, Glunde K. Targeting phospholipid metabolism in cancer. Front Oncol. 2016;6:266. doi:10.3389/fonc.2016.00266. [Google Scholar] [PubMed] [CrossRef]
68. De Molina RA, Gutiérrez R, Ramos MA, Silva JM, Silva J, Bonilla F et al. Increased choline kinase activity in human breast carcinomas: clinical evidence for a potential novel antitumor strategy. Oncogene. 2002;21:4317–22. doi:10.1038/sj.onc.1205556. [Google Scholar] [PubMed] [CrossRef]
69. Iorio E, Mezzanzanica D, Alberti P, Spadaro F, Ramoni C, D’Ascenzo S, et al. Alterations of choline phospholipid metabolism in ovarian tumor progression. Cancer Res. 2005;65:9369–76. doi:10.1158/0008-5472.CAN-05-1146. [Google Scholar] [PubMed] [CrossRef]
70. de Molina RA, Rodríguez-González A, Gutiérrez R, Martínez-Piñeiro L, Sánchez J, Bonilla F et al. Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate, and colorectal human cancers. Biochem Biophys Res Commun. 2002;296:580–3. doi:10.1016/S0006-291X(02)00920-8. [Google Scholar] [PubMed] [CrossRef]
71. Miyake T, Parsons SJ. Functional interactions between Choline kinase α, epidermal growth factor receptor and c-Src in breast cancer cell proliferation. Oncogene. 2012;31:1431–41. doi:10.1038/onc.2011.332. [Google Scholar] [PubMed] [CrossRef]
72. Liu R, Lee J-H, Li J, Yu R, Tan L, Xia Y, et al. Choline kinase alpha 2 acts as a protein kinase to promote lipolysis of lipid droplets. Mol Cell. 2021;81:2722–35. doi:10.1016/j.molcel.2021.05.005. [Google Scholar] [PubMed] [CrossRef]
73. Sanchez-Lopez E, Zimmerman T, Gomez del Pulgar T, Moyer MP, Lacal Sanjuan JC, Cebrian A. Choline kinase inhibition induces exacerbated endoplasmic reticulum stress and triggers apoptosis via CHOP in cancer cells. Cell Death Dis. 2013;4:e933. doi:10.1038/cddis.2013.453. [Google Scholar] [PubMed] [CrossRef]
74. Koch K, Hartmann R, Schröter F, Suwala AK, Maciaczyk D, Krüger AC et al. Reciprocal regulation of the cholinic phenotype and epithelial-mesenchymal transition in glioblastoma cells. Oncotarget. 2016;7:73414–31. doi:10.18632/oncotarget.12337. [Google Scholar] [PubMed] [CrossRef]
75. Lacal JC, Perona R, de Castro J, Cebrián A. Choline Kinase α inhibitors MN58b and RSM932A enhances the antitumor response to cisplatin in lung tumor cells. Pharmaceutics. 2022;14(6):1143. doi:10.3390/pharmaceutics14061143. [Google Scholar] [PubMed] [CrossRef]
76. Chiu D-C, Baskin JM. Imaging and editing the phospholipidome. Acc Chem Res. 2022;55:3088–98. doi:10.1021/acs.accounts.2c00510. [Google Scholar] [PubMed] [CrossRef]
77. Kim HS, Park MY, Yun NJ, Go HS, Kim MY, Seong JK, et al. Targeting PLD2 in adipocytes augments adaptive thermogenesis by improving mitochondrial quality and quantity in mice. J Exp Med. 2022;219:e20211523. doi:10.1084/jem.20211523. [Google Scholar] [PubMed] [CrossRef]
78. Nan Y, Luo Q, Wu X, Liu S, Zhao P, Chang W, et al. DLGAP1-AS2-mediated phosphatidic acid synthesis activates YAP signaling and confers chemoresistance in squamous cell carcinoma. Cancer Res. 2022;82:2887–903. doi:10.1158/0008-5472.CAN-22-0717. [Google Scholar] [PubMed] [CrossRef]
79. Muñoz-Galván S, Lucena-Cacace A, Perez M, Otero-Albiol D, Gomez-Cambronero J, Carnero A. Tumor cell-secreted PLD increases tumor stemness by senescence-mediated communication with microenvironment. Oncogene. 2019;38:1309–23. doi:10.1038/s41388-018-0527-2. [Google Scholar] [PubMed] [CrossRef]
80. Muñoz-Galván S, Verdugo-Sivianes EM, Santos-Pereira JM, Estevez-García P, Carnero A. Essential role of PLD2 in hypoxia-induced stemness and therapy resistance in ovarian tumors. J Exp Clin Cancer Res CR. 2024;43:57. doi:10.1186/s13046-024-02988-y. [Google Scholar] [PubMed] [CrossRef]
81. Henkels KM, Boivin GP, Dudley ES, Berberich SJ, Gomez-Cambronero J. Phospholipase D (PLD) drives cell invasion, tumor growth and metastasis in a human breast cancer xenograph model. Oncogene. 2013;32:5551–62. doi:10.1038/onc.2013.207. [Google Scholar] [PubMed] [CrossRef]
82. Nagumo Y, Kandori S, Tanuma K, Nitta S, Chihara I, Shiga M, et al. PLD1 promotes tumor invasion by regulation of MMP-13 expression via NF-κB signaling in bladder cancer. Cancer Lett. 2021;511:15–25. doi:10.1016/j.canlet.2021.04.014. [Google Scholar] [PubMed] [CrossRef]
83. Kang DW, Hwang WC, Noh YN, Park KS, Min DS. Phospholipase D1 inhibition sensitizes glioblastoma to temozolomide and suppresses its tumorigenicity. J Pathol. 2020;252:304–16. doi:10.1002/path.5519. [Google Scholar] [PubMed] [CrossRef]
84. Lim SH, Lee H, Lee HJ, Kim K, Choi J, Han JM, et al. PLD1 is a key player in cancer stemness and chemoresistance: therapeutic targeting of cross-talk between the PI3K/Akt and Wnt/β-catenin pathways. Exp Mol Med. 2024;56:1479–87. doi:10.1038/s12276-024-01260-9. [Google Scholar] [PubMed] [CrossRef]
85. Chen Y, Zheng Y, Foster DA. Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene. 2003;22:3937–42. doi:10.1038/sj.onc.1206565. [Google Scholar] [PubMed] [CrossRef]
86. Gadiya M, Mori N, Cao MD, Mironchik Y, Kakkad S, Gribbestad IS, et al. Phospholipase D1 and choline kinase-α are interactive targets in breast cancer. Cancer Biol Ther. 2014;15:593–601. doi:10.4161/cbt.28165. [Google Scholar] [PubMed] [CrossRef]
87. Hwang WC, Song D, Lee H, Oh C, Lim SH, Bae HJ, et al. Inhibition of phospholipase D1 induces immunogenic cell death and potentiates cancer immunotherapy in colorectal cancer. Exp Mol Med. 2022;54:1563–76. doi:10.1038/s12276-022-00853-6. [Google Scholar] [PubMed] [CrossRef]
88. Iorio E, Ricci A, Bagnoli M, Pisanu ME, Castellano G, Di Vito M, et al. Activation of phosphatidylcholine cycle enzymes in human epithelial ovarian cancer cells. Cancer Res. 2010;70:2126–35. doi:10.1158/0008-5472.CAN-09-3833. [Google Scholar] [PubMed] [CrossRef]
89. Abalsamo L, Spadaro F, Bozzuto G, Paris L, Cecchetti S, Lugini L, et al. Inhibition of phosphatidylcholine-specific phospholipase C results in loss of mesenchymal traits in metastatic breast cancer cells. Breast Cancer Res BCR. 2012;14:R50. doi:10.1186/bcr3151. [Google Scholar] [PubMed] [CrossRef]
90. S C, I B, R F, L M, S C, F P et al. Inhibition of phosphatidylcholine-specific phospholipase C interferes with proliferation and survival of tumor initiating cells in squamous cell carcinoma. PLoS One. 2015;10:e0136120. doi:10.1371/journal.pone.0136120. [Google Scholar] [PubMed] [CrossRef]
91. Schnoeder TM, Arreba-Tutusaus P, Mohr J, Weinert S, Frey S, Sammt A, et al. Myeloid leukemia stem cells depend on phospholipase C gamma 1 (Plcg1) signaling. Blood. 2016;128:1202–2. doi:10.1182/blood.V128.22.1202.1202. [Google Scholar] [CrossRef]
92. Okada M, Nakagawa-Saito Y, Mitobe Y, Sugai A, Togashi K, Suzuki S, et al. Inhibition of the phospholipase Cε-c-Jun N-terminal kinase axis suppresses glioma stem cell properties. Int J Mol Sci. 2022;23:8785. doi:10.3390/ijms23158785. [Google Scholar] [PubMed] [CrossRef]
93. Sukocheva OA, Hu DG, Meech R, Bishayee A. Divergence of intracellular trafficking of sphingosine Kinase 1 and Sphingosine-1-phosphate receptor 3 in MCF-7 breast cancer cells and MCF-7-derived stem cell-enriched mammospheres. Int J Mol Sci. 2021;22:4314. doi:10.3390/ijms22094314. [Google Scholar] [PubMed] [CrossRef]
94. Yuan H, Zhu B, Li C, Zhao Z. Ceramide in cerebrovascular diseases. Front Cell Neurosci. 2023;17:1191609. doi:10.3389/fncel.2023.1191609. [Google Scholar] [PubMed] [CrossRef]
95. Bataller M, Sánchez-García A, Garcia-Mayea Y, Mir C, Rodriguez I, LLeonart ME. The role of sphingolipids metabolism in cancer drug resistance. Front Oncol. 2021;11:807636. doi:10.3389/fonc.2021.807636. [Google Scholar] [PubMed] [CrossRef]
96. Thayyullathil F, Cheratta AR, Alakkal A, Subburayan K, Pallichankandy S, Hannun YA, et al. Acid sphingomyelinase-dependent autophagic degradation of GPX4 is critical for the execution of ferroptosis. Cell Death Dis. 2021;12:26. doi:10.1038/s41419-020-03297-w. [Google Scholar] [PubMed] [CrossRef]
97. Li C, Klingler S, Bodo S, Cheng J, Pan Y, Adileh M, et al. Acid sphingomyelinase-ceramide induced vascular injury determines colorectal cancer stem cell fate. Cell Physiol Biochem. 2022;56:436–48. doi:10.33594/000000562. [Google Scholar] [PubMed] [CrossRef]
98. Zhong L, Kong JN, Dinkins MB, Leanhart S, Zhu Z, Spassieva SD, et al. Increased liver tumor formation in neutral sphingomyelinase-2-deficient mice. J Lipid Res. 2018;59:795–804. doi:10.1194/jlr.M080879. [Google Scholar] [PubMed] [CrossRef]
99. Lw H, Ff C, Cw M, Zy Y, Hh C, Vj R, et al. Sphingosine Kinase 1 regulates the survival of breast cancer stem cells and non-stem breast cancer cells by suppression of STAT1. Cells. 2020;9:886. doi:10.3390/cells9040886. [Google Scholar] [PubMed] [CrossRef]
100. Pyne NJ, Pyne S. Recent advances in the role of sphingosine 1-phosphate in cancer. FEBS Lett. 2020;594:3583–601. doi:10.1002/1873-3468.13933. [Google Scholar] [PubMed] [CrossRef]
101. Marfia G, Campanella R, Navone SE, Di Vito C, Riccitelli E, Hadi LA, et al. Autocrine/paracrine sphingosine-1-phosphate fuels proliferative and stemness qualities of glioblastoma stem cells. Glia. 2014;62:1968–81. doi:10.1002/glia.22718. [Google Scholar] [PubMed] [CrossRef]
102. Grassi S, Mauri L, Prioni S, Cabitta L, Sonnino S, Prinetti A, et al. Sphingosine 1-phosphate receptors and metabolic enzymes as druggable targets for brain diseases. Front Pharmacol. 2019;10:807. doi:10.3389/fphar.2019.00807. [Google Scholar] [PubMed] [CrossRef]
103. Pedada SR, Yarla NS, Tambade PJ, Dhananjaya BL, Bishayee A, Arunasree KM, et al. Synthesis of new secretory phospholipase A2-inhibitory indole containing isoxazole derivatives as anti-inflammatory and anticancer agents. Eur J Med Chem. 2016;112:289–97. doi:10.1016/j.ejmech.2016.02.025. [Google Scholar] [PubMed] [CrossRef]
104. Yao R-W, Wang Y, Chen L-L. Cellular functions of long noncoding RNAs. Nat Cell Biol. 2019;21:542–51. doi:10.1038/s41556-019-0311-8. [Google Scholar] [PubMed] [CrossRef]
105. Xu W, Xu Q, Kuang D, Wang Z, Lu Q, Lin Q, et al. Long non-coding RNA SLNCR1 regulates non-small cell lung cancer migration, invasion and stemness through interactions with secretory phospholipase A2. Mol Med Rep. 2019;20:2591–6. doi:10.3892/mmr.2019.10518. [Google Scholar] [PubMed] [CrossRef]
106. Liu S, Sun Y, Hou Y, Yang L, Wan X, Qin Y, et al. A novel lncRNA ROPM-mediated lipid metabolism governs breast cancer stem cell properties. J Hematol OncolJ Hematol Oncol. 2021;14:178. doi:10.1186/s13045-021-01194-z. [Google Scholar] [PubMed] [CrossRef]
107. Jiang Y, Guo Y, Hao J, Guenter R, Lathia J, Beck AW, et al. Development of an arteriolar niche and self-renewal of breast cancer stem cells by lysophosphatidic acid/protein kinase D signaling. Commun Biol. 2021;4:780. doi:10.1038/s42003-021-02308-6. [Google Scholar] [PubMed] [CrossRef]
108. Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, et al. FAT SIGNALS–lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012;15:279–91. doi:10.1016/j.cmet.2011.12.018. [Google Scholar] [PubMed] [CrossRef]
109. Reed A, Ichu T-A, Milosevich N, Melillo B, Schafroth MA, Otsuka Y, et al. LPCAT3 inhibitors remodel the polyunsaturated phospholipid content of human cells and protect from ferroptosis. ACS Chem Biol. 2022;17:1607–18. doi:10.1021/acschembio.2c00317. [Google Scholar] [PubMed] [CrossRef]
110. Kopecka J, Trouillas P, Gašparović AČ, Gazzano E, Assaraf YG, Riganti C. Phospholipids and cholesterol: inducers of cancer multidrug resistance and therapeutic targets. Drug Resist Updat Rev Comment Antimicrob Anticancer Chemother. 2020;49:100670. doi:10.1016/j.drup.2019.100670. [Google Scholar] [PubMed] [CrossRef]
111. Lin K-H, Chiang J-C, Chen W-M, Ho Y-H, Yao C-L, Lee H. Transcriptional regulation of lysophosphatidic acid receptors 2 and 3 regulates myeloid commitment of hematopoietic stem cells. Am J Physiol Cell Physiol. 2021;320:C509–19. doi:10.1152/ajpcell.00506.2020. [Google Scholar] [PubMed] [CrossRef]
112. Qiao L, Guo Z, Liu H, Liu J, Lin X, Deng H, et al. Protective effect of mitophagy regulated by mTOR signaling pathway in liver fibrosis associated with selenium. Nutrients. 2022;14:2410. doi:10.3390/nu14122410. [Google Scholar] [PubMed] [CrossRef]
113. Mengzhe Y, Beibei Z, Zhenqiang L, Nannan C, Anqiao L, Jianyu Y, et al. Sanguinarine suppresses cell proliferation, migration and invasion in nasopharyngeal carcinoma inhibiting mTOR signaling. J Tradit Chin Med Chung Tsa Chih Ying Wen Pan. 2022;42:687–92. doi:10.19852/j.cnki.jtcm.20220426.001. [Google Scholar] [PubMed] [CrossRef]
114. Lei W, Yan Y, Ma Y, Jiang M, Zhang B, Zhang H, et al. Notoginsenoside R1 regulates ischemic myocardial lipid metabolism by activating the AKT/mTOR signaling pathway. Front Pharmacol. 2022;13:905092. doi:10.3389/fphar.2022.905092. [Google Scholar] [PubMed] [CrossRef]
115. Kui X, Hua YS, Zhao CM, Guo MM, Wang Y, Yi XJ. GP5 regulates epithelial-mesenchymal transition in breast cancer via the PI3K/AKT signaling pathway. Exp Biol Med Maywood NJ. 2022;247:1501–17. doi:10.1177/15353702221110642. [Google Scholar] [PubMed] [CrossRef]
116. Shen T, Yang T, Yao M, Zheng Z, He M, Shao M, et al. BTC as a novel biomarker contributing to EMT via the PI3K-AKT pathway in OSCC. Front Genet. 2022;13:875617. doi:10.3389/fgene.2022.875617. [Google Scholar] [PubMed] [CrossRef]
117. Su W, Feng B, Hu L, Guo X, Yu M. MUC3A promotes the progression of colorectal cancer through the PI3K/Akt/mTOR pathway. BMC Cancer. 2022;22:602. doi:10.1186/s12885-022-09709-8. [Google Scholar] [PubMed] [CrossRef]
118. Liao J, Chen H, Qi M, Wang J, Wang M. MLLT11-TRIL complex promotes the progression of endometrial cancer through PI3K/AKT/mTOR signaling pathway. Cancer Biol Ther. 2022;23:211–24. doi:10.1080/15384047.2022.2046450. [Google Scholar] [PubMed] [CrossRef]
119. Wang Y, Luo S, Wang Y, Yang S, Huang Z, Zhu X et al. EID3 promotes cancer stem cell-like phenotypes in osteosarcoma through the activation of PI3K-AKT signaling pathway. Oxid Med Cell Longev. 2022;2022:5941562. doi:10.1155/2022/5941562. [Google Scholar] [PubMed] [CrossRef]
120. Mangiapane LR, Nicotra A, Turdo A, Gaggianesi M, Bianca P, Di Franco S, et al. PI3K-driven HER2 expression is a potential therapeutic target in colorectal cancer stem cells. Gut. 2022;71:119–28. doi:10.1136/gutjnl-2020-323553. [Google Scholar] [PubMed] [CrossRef]
121. Zhang Y, Tseng C-C, Tsai Y-L, Fu X, Schiff R, Lee AS. Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PLoS One. 2013;8:e80071. doi:10.1371/journal.pone.0080071. [Google Scholar] [PubMed] [CrossRef]
122. Álvarez-Garcia V, Tawil Y, Wise HM, Leslie NR. Mechanisms of PTEN loss in cancer: it’s all about diversity. Semin Cancer Biol. 2019;59:66–79. doi:10.1016/j.semcancer.2019.02.001. [Google Scholar] [PubMed] [CrossRef]
123. Liu J, Chen W, Zhang H, Liu T, Zhao L. miR-214 targets the PTEN-mediated PI3K/Akt signaling pathway and regulates cell proliferation and apoptosis in ovarian cancer. Oncol Lett. 2017;14:5711–8. doi:10.3892/ol.2017.6953. [Google Scholar] [PubMed] [CrossRef]
124. Al-Dhfyan A, Alhoshani A, Korashy HM. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and β-Catenin and Akt activation. Mol Cancer. 2017;16:14. doi:10.1186/s12943-016-0570-y. [Google Scholar] [PubMed] [CrossRef]
125. Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira S-M, García-Echeverría C, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci USA. 2009;106:268–73. doi:10.1073/pnas.0810956106. [Google Scholar] [PubMed] [CrossRef]
126. Lucaciu A, Brunkhorst R, Pfeilschifter JM, Pfeilschifter W, Subburayalu J. The S1P-S1PR axis in neurological disorders-insights into current and future therapeutic perspectives. Cells. 2020;9:E1515. doi:10.3390/cells9061515. [Google Scholar] [PubMed] [CrossRef]
127. Cheng J, Ma H, Yan M, Zhang Z, Xing W. Circ_0007624 suppresses the development of esophageal squamous cell carcinoma via targeting miR-224-5p/CPEB3 to inactivate the EGFR/PI3K/AKT signaling. Cell Signal. 2022;99:110448. doi:10.1016/j.cellsig.2022.110448. [Google Scholar] [PubMed] [CrossRef]
128. Kim S, You D, Jeong Y, Yoon SY, Lo E, Lee JE. Celastrol attenuates amphiregulin expression by inhibiting MAPK signaling pathway in triple-negative breast cancer cells. Phytomedicine Plus. 2022;2:100319. doi:10.1016/j.phyplu.2022.100319. [Google Scholar] [CrossRef]
129. Kim S, Cho H, Hong S-O, Oh SJ, Lee H-J, Cho E, et al. LC3B upregulation by NANOG promotes immune resistance and stem-like property through hyperactivation of EGFR signaling in immune-refractory tumor cells. Autophagy. 2021;17:1978–97. doi:10.1080/15548627.2020.1805214. [Google Scholar] [PubMed] [CrossRef]
130. Maeda R, Tamagaki-Asahina H, Sato T, Yanagawa M, Sako Y. Threonine phosphorylation regulates the molecular assembly and signaling of EGFR in cooperation with membrane lipids. J Cell Sci. 2022;135:jcs260355. doi:10.1242/jcs.260355. [Google Scholar] [PubMed] [CrossRef]
131. Li X, Wang H, Yang X, Wang X, Zhao L, Zou L, et al. GABRP sustains the stemness of triple-negative breast cancer cells through EGFR signaling. Cancer Lett. 2021;514:90–102. doi:10.1016/j.canlet.2021.04.028. [Google Scholar] [PubMed] [CrossRef]
132. Gan HK, Cvrljevic AN, Johns TG. The epidermal growth factor receptor variant III (EGFRvIIIwhere wild things are altered. FEBS J. 2013;280:5350–70. doi:10.1111/febs.12393. [Google Scholar] [PubMed] [CrossRef]
133. Bi J, Ichu T-A, Zanca C, Yang H, Zhang W, Gu Y, et al. Oncogene amplification in growth factor signaling pathways renders cancers dependent on membrane lipid remodeling. Cell Metab. 2019;30:525–38. doi:10.1016/j.cmet.2019.06.014. [Google Scholar] [PubMed] [CrossRef]
134. Liu F, Wu Y, Liu J, Ni R-J, Yang A-G, Bian K, et al. A miR-205-LPCAT1 axis contributes to proliferation and progression in multiple cancers. Biochem Biophys Res Commun. 2020;527:474–80. doi:10.1016/j.bbrc.2020.04.071. [Google Scholar] [PubMed] [CrossRef]
135. Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, Spellman PT, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1:84–96. doi:10.1016/j.molonc.2007.02.004. [Google Scholar] [PubMed] [CrossRef]
136. Zhang D, LaFortune TA, Krishnamurthy S, Esteva FJ, Cristofanilli M, Liu P, et al. Epidermal growth factor receptor tyrosine kinase inhibitor reverses mesenchymal to epithelial phenotype and inhibits metastasis in inflammatory breast cancer. Clin Cancer Res off J Am Assoc Cancer Res. 2009;15:6639–48. doi:10.1158/1078-0432.CCR-09-0951. [Google Scholar] [PubMed] [CrossRef]
137. Li Y-W, Xu J, Zhu G-Y, Huang Z-J, Lu Y, Li X-Q, et al. Apigenin suppresses the stem cell-like properties of triple-negative breast cancer cells by inhibiting YAP/TAZ activity. Cell Death Discov. 2018;4:105. doi:10.1038/s41420-018-0124-8. [Google Scholar] [PubMed] [CrossRef]
138. Ghasemi H, Mousavibahar SH, Hashemnia M, Karimi J, Khodadadi I, Mirzaei F, et al. Tissue stiffness contributes to YAP activation in bladder cancer patients undergoing transurethral resection. Ann N Y Acad Sci. 2020;1473:48–61. doi:10.1111/nyas.14358. [Google Scholar] [PubMed] [CrossRef]
139. Wang K-J, Wang C, Dai L-H, Yang J, Huang H, Ma X-J, et al. Targeting an autocrine regulatory loop in cancer stem-like cells impairs the progression and chemotherapy resistance of bladder cancer. Clin Cancer Res off J Am Assoc Cancer Res. 2019;25:1070–86. doi:10.1158/1078-0432.CCR-18-0586. [Google Scholar] [PubMed] [CrossRef]
140. Thirusangu P, Ray U, Sarkar Bhattacharya S, Oien DB, Jin L, Staub J, et al. PFKFB3 regulates cancer stemness through the hippo pathway in small cell lung carcinoma. Oncogene. 2022;41:4003–17. doi:10.1038/s41388-022-02391-x. [Google Scholar] [PubMed] [CrossRef]
141. Liang Y, Voshart D, Paridaen JTML, Oosterhof N, Liang D, Thiruvalluvan A, et al. CD146 increases stemness and aggressiveness in glioblastoma and activates YAP signaling. Cell Mol Life Sci. 2022;79:398. doi:10.1007/s00018-022-04420-0. [Google Scholar] [PubMed] [CrossRef]
142. Yu F-X, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780–91. doi:10.1016/j.cell.2012.06.037. [Google Scholar] [PubMed] [CrossRef]
143. Han H, Qi R, Zhou JJ, Ta AP, Yang B, Nakaoka HJ, et al. Regulation of the Hippo pathway by phosphatidic acid-mediated lipid-protein interaction. Mol Cell. 2018;72:328–40. doi:10.1016/j.molcel.2018.08.038. [Google Scholar] [PubMed] [CrossRef]
144. Lin L, Wen J, Lin B, Chen H, Bhandari A, Qi Y, et al. Phospholipase C Delta 3 inhibits apoptosis and promotes proliferation, migration, and invasion of thyroid cancer cells via Hippo pathway. Acta Biochim Biophys Sin. 2021;53:481–91. doi:10.1093/abbs/gmab016. [Google Scholar] [PubMed] [CrossRef]
145. Kemppainen K, Wentus N, Lassila T, Laiho A, Törnquist K. Sphingosylphosphorylcholine regulates the Hippo signaling pathway in a dual manner. Cell Signal. 2016;28:1894–903. doi:10.1016/j.cellsig.2016.09.004. [Google Scholar] [PubMed] [CrossRef]
146. Noto A, De Vitis C, Pisanu ME, Roscilli G, Ricci G, Catizone A, et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene. 2017;36:4573–84. doi:10.1038/onc.2017.75. [Google Scholar] [PubMed] [CrossRef]
147. Hu C-B, Jiang H, Yang Y, Wang G-H, Ji Q-H, Jia Z-Z, et al. DL-3-n-butylphthalide alleviates motor disturbance by suppressing ferroptosis in a rat model of Parkinson’s disease. Neural Regen Res. 2023;18:194–9. doi:10.4103/1673-5374.343892. [Google Scholar] [PubMed] [CrossRef]
148. Shen D, Luo J, Chen L, Ma W, Mao X, Zhang Y, et al. PARPi treatment enhances radiotherapy-induced ferroptosis and antitumor immune responses via the cGAS signaling pathway in colorectal cancer. Cancer Lett. 2022;550:215919. doi:10.1016/j.canlet.2022.215919. [Google Scholar] [PubMed] [CrossRef]
149. Yang K, Zeng L, Yuan X, Wang S, Ge A, Xu H, et al. The mechanism of ferroptosis regulating oxidative stress in ischemic stroke and the regulation mechanism of natural pharmacological active components. Biomed Pharmacother. 2022;154:113611. doi:10.1016/j.biopha.2022.113611. [Google Scholar] [PubMed] [CrossRef]
150. Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc− and thereby promotes tumor growth. Cancer Cell. 2011;19:387–400. doi:10.1016/j.ccr.2011.01.038. [Google Scholar] [PubMed] [CrossRef]
151. Liu T, Jiang L, Tavana O, Gu W. The Deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res. 2019;79:1913–24. doi:10.1158/0008-5472.CAN-18-3037. [Google Scholar] [PubMed] [CrossRef]
152. Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90. doi:10.1038/nchembio.2238. [Google Scholar] [PubMed] [CrossRef]
153. Cui S, Simmons G, Vale G, Deng Y, Kim J, Kim H, et al. FAF1 blocks ferroptosis by inhibiting peroxidation of polyunsaturated fatty acids. Proc Natl Acad Sci U S A. 2022;119:e2107189119. doi:10.1073/pnas.2107189119. [Google Scholar] [PubMed] [CrossRef]
154. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–3. doi:10.1038/nature07733. [Google Scholar] [PubMed] [CrossRef]
155. Arnold CR, Mangesius J, Skvortsova I-I, Ganswindt U. The role of cancer stem cells in radiation resistance. Front Oncol. 2020;10:164. doi:10.3389/fonc.2020.00164. [Google Scholar] [PubMed] [CrossRef]
156. Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, et al. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell. 2015;28:441–55. doi:10.1016/j.ccell.2015.09.002. [Google Scholar] [PubMed] [CrossRef]
157. Taylor WR, Fedorka SR, Gad I, Shah R, Alqahtani HD, Koranne R, et al. Small-molecule ferroptotic agents with potential to selectively target cancer stem cells. Sci Rep. 2019;9:5926. doi:10.1038/s41598-019-42251-5. [Google Scholar] [PubMed] [CrossRef]
158. Elgendy SM, Alyammahi SK, Alhamad DW, Abdin SM, Omar HA. Ferroptosis: an emerging approach for targeting cancer stem cells and drug resistance. Crit Rev Oncol Hematol. 2020;155:103095. doi:10.1016/j.critrevonc.2020.103095. [Google Scholar] [PubMed] [CrossRef]
159. Polonio-Alcalá E, Porta R, Ruiz-Martínez S, Vásquez-Dongo C, Relat J, Bosch-Barrera J, et al. AZ12756122, a novel fatty acid synthase inhibitor, decreases resistance features in EGFR-TKI resistant EGFR-mutated NSCLC cell models. Biomed Pharmacother. 2022;156:113942. doi:10.1016/j.biopha.2022.113942. [Google Scholar] [PubMed] [CrossRef]
160. Yu Y, Kim H, Choi S, Yu J, Lee JY, Lee H, et al. Targeting a lipid desaturation enzyme, SCD1, selectively eliminates colon cancer stem cells through the suppression of Wnt and NOTCH signaling. Cells. 2021;10:106. doi:10.3390/cells10010106. [Google Scholar] [PubMed] [CrossRef]
161. Tracz-Gaszewska Z, Dobrzyn P. Stearoyl-CoA desaturase 1 as a therapeutic target for the treatment of cancer. Cancers. 2019;11:948. doi:10.3390/cancers11070948. [Google Scholar] [PubMed] [CrossRef]
162. Kim SJ, Park SJ, Park J, Cho HJ, Shim J-K, Seon J, et al. Dual inhibition of CPT1A and G6PD suppresses glioblastoma tumorspheres. J Neurooncol. 2022;160:677–89. doi:10.1007/s11060-022-04189-z. [Google Scholar] [PubMed] [CrossRef]
163. Nakayama S, Yoda E, Yamashita S, et al. Knockdown of iPLA2γ enhances cisplatin-induced apoptosis by increasing ROS-dependent peroxidation of mitochondrial phospholipids in bladder cancer cells. Free Radic Biol Med. 2024;220:301–11. doi:10.1016/j.freeradbiomed.2024.05.016. [Google Scholar] [PubMed] [CrossRef]
164. Wang G, Xu J, Zhao J, Yin W, Liu D, Chen W, et al. Arf1-mediated lipid metabolism sustains cancer cells and its ablation induces anti-tumor immune responses in mice. Nat Commun. 2020;11:220. doi:10.1038/s41467-019-14046-9. [Google Scholar] [PubMed] [CrossRef]
165. Wen D, Wang J, Van Den Driessche G, Chen Q, Zhang Y, Chen G, et al. Adipocytes as anticancer drug delivery depot. Matter. 2019;1:1203–14. doi:10.1016/j.matt.2019.08.007. [Google Scholar] [CrossRef]
166. Du Y, Wang S, Zhang M, Chen B, Shen Y. Cells-based drug delivery for cancer applications. Nanoscale Res Lett. 2021;16:139. doi:10.1186/s11671-021-03588-x. [Google Scholar] [PubMed] [CrossRef]
167. Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: at the bench and bedside. Semin Cancer Biol. 2019;59:125–32. doi:10.1016/j.semcancer.2019.07.009. [Google Scholar] [PubMed] [CrossRef]
168. Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. 2021;11:1913–22. doi:10.1158/2159-8290.CD-21-0365. [Google Scholar] [PubMed] [CrossRef]
169. Kirtonia A, Sethi G, Garg M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell Mol Life Sci. 2020;77:4459–83. doi:10.1007/s00018-020-03536-5. [Google Scholar] [PubMed] [CrossRef]
170. Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med. 2020;52(2):192–203. doi:10.1038/s12276-020-0384-2. [Google Scholar] [PubMed] [CrossRef]
171. Zhu Y, Archer WR, Morales KF, Schulz MD, Wang Y, Matson JB. Enzyme-triggered chemodynamic therapy via a peptide-H2S donor conjugate with complexed Fe2+. Angew Chem Int Ed Engl. 2023;62:e202302303. doi:10.1002/anie.202302303. [Google Scholar] [PubMed] [CrossRef]
172. Zhang S-L, Liu C, Li Z-X, Guan Y-H, Ge L, Sun Q, et al. Sonoactivated cascade fenton reaction enhanced by synergistic modulation of electron-hole separation for improved tumor therapy. Adv Healthc Mater. 2023;12:2300982. doi:10.1002/adhm.202300982. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools