
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW
Epigenetics of Malignant Melanoma: Mechanisms, Diagnostic Approaches and Therapeutic Applications
1 Department of Human Developmental and Regenerative Biology, Harvard University, Cambridge, MA 02138, USA
2 Program in Dermatopathology, Department of Pathology, Brigham and Women’s Hospital, Mass General Brigham, Harvard Medical School, Boston, MA 02115, USA
* Corresponding Author: Christine G. Lian. Email:
(This article belongs to the Special Issue: Advances in Skin Cancer Management: From Molecular Targets to Innovative Treatments)
Oncology Research 2026, 34(4), 4 https://doi.org/10.32604/or.2026.073894
Received 28 September 2025; Accepted 19 December 2025; Issue published 23 March 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Malignant melanoma (MM) is a highly aggressive skin cancer known for its rapid progression, potential for metastasis, and resistance to treatment. Despite advances in targeted therapies and immunotherapy, the prognosis for metastatic melanoma remains unfavorable. Recent research has shed light on the significance of epigenetic modifications in the pathogenesis of melanoma, revealing critical mechanisms of melanoma development and progression. Epigenetic modifications, including DNA and RNA modifications, histone modifications, chromatin remodeling, and non-coding RNA regulation, disrupt normal gene expression without modifying the DNA sequence, leading to cellular transformation, invasion, immune evasion, and therapeutic resistance. The reversible nature of epigenetic modifications opens up new opportunities for melanoma recognition and classification, as well as therapeutic applications, including the development of diagnostic and prognostic biomarkers and innovative targeted therapies aimed at restoring normal gene function and enhancing the efficacy of existing treatments. This review will focus on the multifaceted role of epigenetic dysregulation in melanoma. The future integration of epigenetic data and genomic profiling with clinical outcomes, likely facilitated by artificial intelligence (AI) algorithms, holds promise for personalized treatment strategies that are informed by precise and combinatorial diagnostic tools, ultimately improving melanoma care. The study aims to deliver a comprehensive overview of the current state of epigenetics in melanoma.Keywords
1 Epigenetic Mechanisms in Melanoma Pathogenesis
Epigenetics involves modifications in gene expression that may be heritable but are without DNA sequence alterations [1]. The mechanisms of epigenetic aberration include DNA [2], histone [3], and RNA modification [4]; chromatin remodeling [5]; and non-coding RNA regulation [6]. The proteins involved in epigenetic modifications have recently been functionally categorized as writers, erasers, readers, and remodelers [7,8]. Writers and erasers are enzymes that have opposite effects on gene expression, as erasers undo what writers have modified on DNA bases or amino acids. Readers are protein domains that bind to DNA modification sites to recruit other enzymes for gene regulation. Remodelers affect chromatin by modifying chromatin accessibility via attaching or detaching nucleosomes at enhancer and promoter sites [9].
In melanoma, these epigenetic processes are frequently dysregulated and have been recognized as critical factors in cancer development, metastasis, and treatment resistance [10]. The reversibility of the enzymes that catalyze the epigenetic reactions opens up vast possibilities for innovation in drug targeting. We will discuss the epigenetic mechanisms of melanoma pathogenesis in the following order: DNA modification, histone modification, non-coding RNA regulation, chromatin remodeling, and RNA modification. Fig. 1 illustrates the overview of the key mechanisms of epigenetic modifications in melanoma.
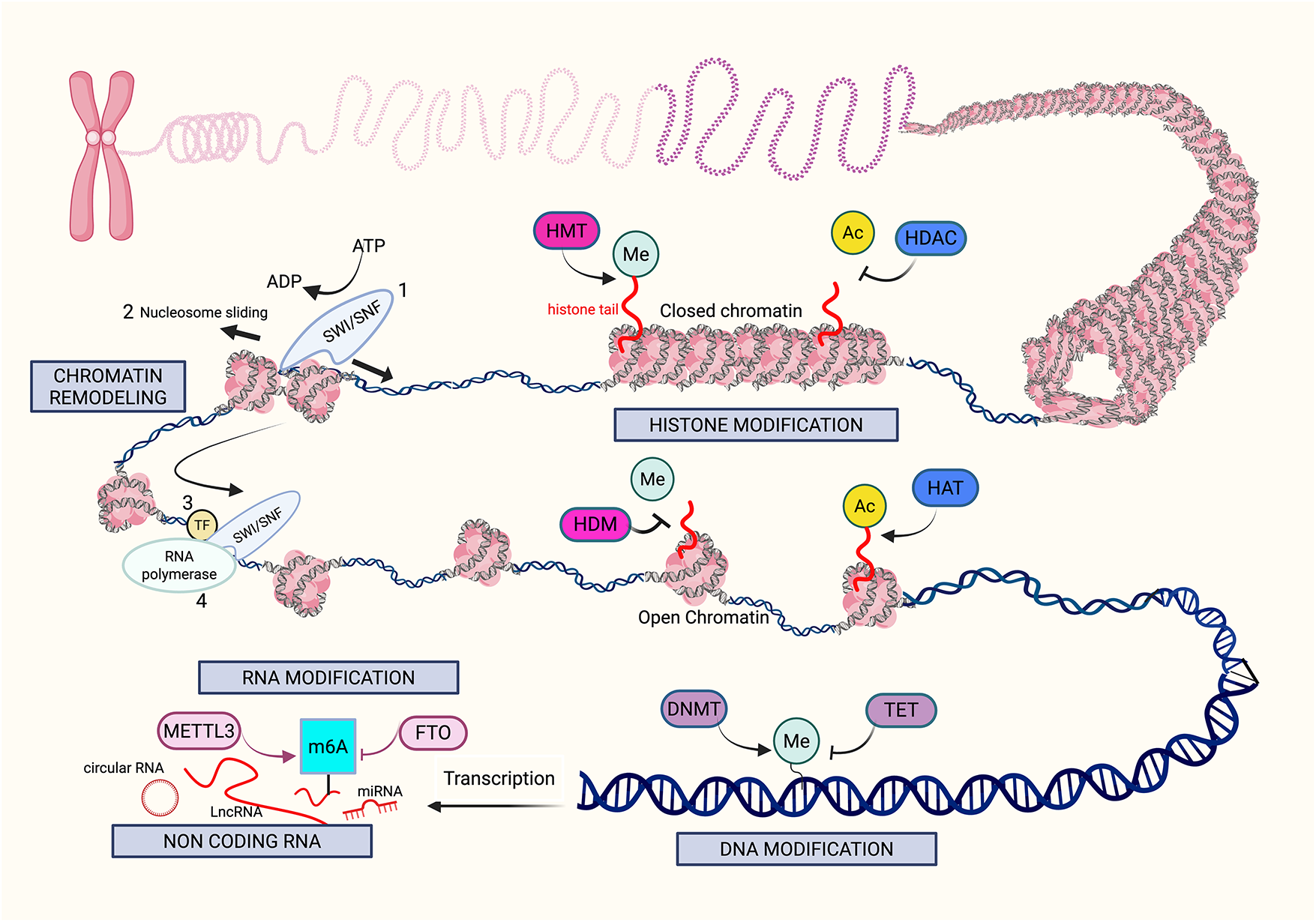
Figure 1: Mechanisms of epigenetic modifications in melanoma. The figure provides an overview of the key mechanisms (gray boxes) that drive epigenetic alterations in melanoma, including histone modification, chromatin remodeling, DNA modification, RNA modification, and noncoding RNA. Histone modifications, such as methylation (Me) and acetylation (Ac), are performed by “writers” like histone methyltransferases (HMT) and histone acetyltransferases (HAT). In contrast, histone deacetylases (HDAC) and histone demethylases (HDM) function as “erasers,” removing these modifications. Histone methylation can be associated with both open and closed chromatin structures, and histone acetylation typically promotes a more open chromatin structure, allowing greater access for transcription factors. Chromatin remodeling initiates with SWItch/Sucrose Non-Fermenting (SWI/SNF) complexes, disrupting histone-DNA interactions by mobilizing nucleosomes, thereby increasing DNA accessibility, which facilitates the binding of transcription factors (TFs) and RNA polymerase to promoter and enhancer regions of the DNA. DNA modifications are mediated by “writers” such as DNA methyltransferases (DNMT) and “erasers” such as Ten-Eleven Translocation (TET) enzymes. An example of RNA modification includes Methyltransferase-like 3 (METTL3), which is a N6-methyladenosine (m6A) “writer”, as it adds methyl groups to adenosine bases on RNA, specifically m6A. Demethylation by Fat mass and obesity-associated protein (FTO) is the “eraser” of this reaction, removing the m6A from the RNA. Lastly, noncoding RNAs such as long noncoding RNAs and microRNAs possess complex epigenetic functions that can alter melanoma pathogenesis. Created in BioRender. Hong, S. (2025)
1.1 DNA Modification in Melanoma
DNA methylation involves the covalent transfer of a methyl group to the 5′ position of cytosine, producing 5-methylcytosine (5mC). Methylation most commonly occurs at the Cytosine-phosphodiester bond-Guanine (CpG) dinucleotides concentrated at the gene promoter region, thus forming “CG islands” [11]. The writer enzymes in DNA methylation are DNA methyltransferases (DNMTs) [12,13], and the eraser enzymes are the ten-eleven translocation (TET) methylcytosine dioxygenases. Hydroxymethylation of 5mC converts it into an intermediary, 5-hydroxymethylcytosine (5-hmC) [14,15], which is interestingly known to be present at elevated levels in self-renewing and pluripotent embryonic stem cells [15,16].
One common mechanism of aberrant DNA methylation in the development of cancer involves hypermethylation of CG islands (CGIs) in gene promoters and global loss of DNA methylation. DNA hypermethylation of tumor suppressor and DNA repair genes impairs the proliferation and differentiation of normal cells, while promoting the development of cancer cells [17–20]. Global loss of DNA methylation causes chromosomal instability [21,22]. Additionally, specific loss of DNA methylation in oncogene promoter regions will also promote malignancy [23]. Various mechanisms of altered DNA methylation play complex roles in cancer development and progression in melanoma, contributing to the loss of cell cycle regulation, apoptosis, proliferation, cell invasion, and metastasis [24]. Global hypomethylation of DNA is a key feature of cancer, marked by a general reduction in 5-methylcytosine throughout the genome [25,26], which encompasses the intergenic region and is present within the introns of DNA, especially in repetitive sequences and transposable elements [27]. Some of these globally hypomethylated repetitive sequences are LINE1, Alu, and Satα [28,29]. Global DNA hypomethylation has been linked to the initial phases of cancer development in diverse cancer types [30,31]. There are indications that hypomethylation may play a role in cancer development by activating oncogenes and causing chromosome instability since methylation of pericentromeric areas is essential for maintaining chromosome stability [23].
In malignant melanomas, global hypomethylation has been observed [32].Compared to nevi, a decrease in 5mC levels is noted in melanomas, which indicates that DNA hypomethylation may have a significant role in the survival of melanoma cells [33]. In addition to global changes, hypomethylation of specific regions/genes has been studied. Hypomethylation activates certain Cancer Testis (CT) genes [34–36], including Melanoma Antigen Genes (MAGE) [37], B Melanoma Antigen (BAGE), G Antigen (GAGE), and New York-Esophageal Squamous cell carcinoma-1 (NY-ESO-1) [38,39]. Other genes with regional hypomethylation include the feline sarcoma (FES) [40], Deleted split-hand/split foot 1 (DSS1) [41], Carnosine dipeptidase 1 (CNDP1) [42], and Tre-2/Bub2/Cdc16-1 Domain Family Member 16 (TBC1D16) [43].
We reported that loss of 5-hmC is an epigenetic hallmark with important functional, diagnostic, and prognostic implications in melanoma [44]. We found that decreased activity of Isocitrate Dehydrogenase 2 (IDH2) and TET family enzymes represents critical mechanisms responsible for the reduction of 5-hmC in melanoma [44]. Of interest, alterations in the genes IDH1 and IDH2 in cancer cells lead to the formation of the oncometabolite 2-hydroxyglutarate (2-HG) [45]. This compound acts as an antagonist to α-ketoglutarate (α-KG), which is essential for the conversion of 5mC to 5-hmC [46]. In animal models, we learned that reintroducing active TET2 or IDH2 in melanoma cells was effective in reducing melanoma growth and extending tumor-free survival. This research highlights the crucial role of 5-hmC in the development and virulence of melanoma, establishing a direct connection between the IDH and TET activity-dependent epigenetic mechanisms and the suppression of melanoma progression through 5-hmC [44]. Moreover, we and others have also found loss of 5-hmC 1) in fields of presumed precursor melanocytes in which melanomas arose [47–50]; 2) in dysplastic nevus precursors in which some melanomas developed [51]; 3) in association with melanoma progression and metastatic process [33,48,52,53]. Epigenetic modifications including the progressive loss of 5-hmC are considered a key component of melanocyte field cancerization [49,50].
We recently examined the expression of Preferentially Expressed Antigen in Melanoma (PRAME) and found that 5-hmC, in conjunction with the activation of PRAME, contributes significantly to the transformation and progression of melanoma. The study revealed that TET2 is significantly less active in melanoma compared to melanocytes found in normal skin, and that this lower activity in melanoma is associated with decreased levels of 5-hmC and increased expression of PRAME [54]. Notably, levels of 5-hmC were reduced at the PRAME 5′ promoter in melanoma compared with nevi, suggesting a role for 5-hmC in PRAME transcription. Restoration of 5-hmC levels via TET2 overexpression in melanoma cell lines markedly reduced PRAME expression, thus establishing a function of TET2-mediated DNA hydroxymethylation in regulating PRAME expression and demonstrating that epigenetic reprogramming plays a potentially pivotal role in melanoma tumorigenesis.
Since Horvath first introduced the concept of universal epigenetic clock for assessing biological age, subsequent research has shown that cancers often exhibit DNA methylation patterns that indicate they are biologically older than healthy tissue, a phenomenon known as “age acceleration” [55]. Aberrant DNA methylation in melanoma dysregulates various cellular processes, including cell cycle, cell signaling, transcription, DNA repair, and apoptosis [35,56–58]. DNA methylation is also found to be associated with the progression of metastatic melanoma. When the metastasis driver, Nuclear Receptor Subfamily 2 Group F, Member 2-isoform 2 (NR2F2-Iso2) is hypomethylated and re-expressed, it enables melanocytes to acquire features similar to those of neural crest cells during metastasis [59]. Table A1 categorizes the genes with aberrant DNA methylation based on the predominant pathogenic mechanism(s) in melanoma evolution and progression.
Phosphatase and TENsin homolog (PTEN) acts as a lipid phosphatase that converts phosphatidylinositol (3,4,5)-triphosphate (PIP3) to PIP2, thereby directly antagonizing phosphatidylinositol 3-kinase (PI3K) signaling. PI3K activation drives hyperactivation of the Ak strain transforming/mammalian target of rapamycin (AKT/mTOR) pathway [60]. This hyperactivation promotes cell proliferation, survival, invasion, and immune evasion through mechanisms such as enhanced Fos-related antigen 1 (FRA1) translation, inactivation of Forkhead box O (FOXO), and increased Programmed Death-Ligand 1 (PD-L1) expression [61]. Hypermethylation has been found to be a significant mechanism of PTEN loss in 60% of melanomas in one study [62,63], although another study found that methylation is less critical, as PTEN is often inactivated through DNA mutations or deletions. However, when PTEN mRNA expression and methylation were plotted and compared, 97.9% of melanomas showed less than 10% PTEN methylation [64]. Cyclin-Dependent Kinase Inhibitor 2A (CDKN2A) encodes two tumor suppressors: protein16 inhibitor of CDK4/6 (p16INK4a), which inhibits CDK4/6 to prevent retinoblastoma tumor suppressor gene (RB) phosphorylation and cell cycle progression, and p14 Alternate Reading Frame (p14ARF), which stabilizes p53 by sequestering murine double minute 2 (MDM2). Loss of CDKN2A through deletion, mutation, or methylation results in uncontrolled E2F activity, directly activating the Brain-2 (BRN2) invasion program and simultaneously eliminating both RB and p53 tumor suppressor checkpoints [65]. Methylation of the CDKN2A gene in melanoma is a key example of a writer alteration in DNA methylation. The methylation leads to the loss of its tumor-suppressive functions, promoting the development of melanoma, and correlating with a poor prognosis. The CDKN2A p16 is hypermethylated in certain cutaneous melanomas, leading to cell cycle arrest at the G1-S checkpoint by inhibiting the proteins CDK4/6 [66–69]. P14ARF (CDKN2A) is found to be hypermethylated in cutaneous and uveal melanomas. One study of melanomas in the vertical growth phase showed that 19% of the cases had CDKN2A promoter region hypermethylation, and interestingly, some instances were heterogeneous with tumor cells that were both methylated and unmethylated [65,68,70]. Ras association domain family 1 isoform A (RASSF1A) may exhibit hypermethylation at its promoter regions. This hypermethylation has been identified in 55% of melanoma tumors, while normal skin shows no detectable methylation. The hypermethylation leads to a halt in the cell cycle (G1 to S phase) and increases the expression of ASK1 (Apoptosis signal-regulating kinase 1), a protein kinase involved in stress-induced cellular responses, particularly apoptosis (programmed cell death) and inflammation. The extent of RASSF1A methylation differs according to the stage of the tumor, and its reduced expression inhibits apoptosis [71,72].
Telomerase Reverse Transcriptase (TERT) promoter-activating mutations were initially identified at a high rate in cutaneous melanoma [73,74], and subsequent studies have indicated their presence in various other cancers [75,76]. Upregulation of TERT plays a role in maintaining telomeres, which is vital for cellular immortality and the survival of cancer cells. While TERT promoter mutations (TPMs) significantly contribute to TERT upregulation in cancer, many tumors show TERT upregulation without these mutations. Research has identified the TERT hypermethylated oncological region (THOR), situated just upstream of the TERT core promoter, as an epigenetic site associated with TERT upregulation in cancer. When THOR is unmethylated, it inhibits TERT promoter activity, regardless of the presence of TPM, while hypermethylation of THOR reverses this inhibitory effect. Thus, THOR hypermethylation is suggested to be a common mechanism for activating telomerase in cancer that can work independently or in collaboration with TPMs [77]. However, the effect of TERT epigenetic regulation on melanoma progression has shown discrepant findings. A study of normal skin samples and 61 melanoma cell lines was conducted to clarify the effects of epigenetic and genetic mechanisms in regulating TERT gene expression. TERT gene expression requires a high promoter methylation level, open chromatin, and the absence of mutations. The TERT gene is also expressed with a moderately methylated promoter and existing mutations. Thus, there is a complex interplay between the promoter methylation, chromatin accessibility, and promoter mutation status [78]. TERT promoter methylation was found to indicate worse prognosis in young melanoma patients, as the promoter methylation alone or combined with promoter mutations correlated with reduced recurrence-free survival, whereas only having the TERT promoter mutation did not correlate with prognosis [79]. O6-methylguanine-DNA methyltransferase (MGMT) is an enzyme that repairs the impact of methylation on DNA, a process that removes alkyl groups from the O6 position of guanine. In melanoma, it is common to find hypermethylation of the MGMT promoter, leading to the silencing of the MGMT gene. This silencing renders the melanoma cells more susceptible to specific chemotherapeutic drugs that utilize alkylating agents to induce DNA damage, leading to improved treatment outcomes. Importantly, the methylation status of MGMT may also serve as a prognostic marker, influencing treatment choices [80,81].
1.2 Histone Modification in Melanoma
Chromatin consists of building blocks of nucleosomes, which consist of DNA (146 base pairs) organized around a histone protein octamer (2 copies of H2A, H2B, H3, H4) [82,83]. Histone modification is a dynamic process influenced by specific enzymes that alter charges determined by nucleosomal structure. This modification either strengthens or weakens interactions between histones and DNA that regulate transcriptional activation and repression [84,85]. Histone modifications primarily occur on the lysine-rich N-terminal tails [86] and cause abnormalities in chromatin structure, influencing gene expression related to cell differentiation, proliferation, and survival. Abnormal histone modifications such as acetylation, phosphorylation, methylation, and ubiquitination significantly contribute to melanoma development by activating oncogenes and silencing tumor suppressor genes. The most common types of epigenetic modifications in melanoma involve the histone acetyl and methyl groups.
1.2.1 Histone Acetylation and Deacetylation
Histone Acetyltransferases (HATs): Writers
The positive charge of histone tails and the negative charge of DNA form a tight bond inherent in the closed heterochromatin structure, and HATs neutralize this histone positive charge to increase chromatin accessibility, thus enhancing transcription [87,88]. Protein 300/CREB-binding protein (P300/CBP) is a biomarker protein produced by histone 3 at lysine 27 (H3K27Ac) acetylation that is neutralized by HAT. Protein 300 and CBP are HATs vital in regulating chromatin dynamics and gene expression. They are involved in various cellular functions, including proliferation, differentiation, and immune responses. Disruption of p300/CBP function has been associated with the onset and progression of melanoma, which includes activating oncogenic transcription factors such as Microphthalmia-Associated Transcription Factor (MITF) and Sex-determining region Y (SRY)-related High-Mobility group (HMG) box (SOX)10, as well as modulating cell cycle progression. Indeed, inhibiting p300/CBP can decrease melanoma cell proliferation and alter gene expression related to melanoma development [89–92].
Bromodomain and Extra-Terminal Domain (BET) Proteins: Readers
BET proteins, specifically Bromodomain-Containing Protein 2 (BRD2) and BRD4, function as readers, and they are both upregulated by acetylation of lysine residues of histones. In melanoma, these proteins are often up-regulated and play a role in tumorigenesis by modulating the expression of essential cell cycle and survival genes. When BRD2 and BRD4 are displaced from chromatin, transcription is inhibited, leading to the deactivation of cell cycle genes such as Extracellular signal-Regulated Kinase 1 (ERK1), cellular myelocytomatosis oncogene (c-MYC), and S-phase kinase-interacting protein 2 (SPK2). This results in G1/S phase arrest and cell death [93]. Segura et al. found that BRD4 levels are elevated in both primary and metastatic melanoma compared to normal melanocytes and melanocytic nevi. The use of BET inhibitors has been shown to hinder melanoma cell growth in vitro as well as impede tumor growth and metastatic activity in vivo. Notably, the effectiveness of these inhibitors is not dependent on the mutational status of B-Raf proto-oncogene, serine/threonine kinase (BRAF) or neuroblastoma RAS viral oncogene homolog (NRAS), suggesting that these small-molecule therapies could represent viable treatment options [94].
Histone Deacetylases (HDACs): Erasers
HDACs—such as HDAC6 [95,96], HDAC1 [97], HDAC3 [98] and HDAC8 [98]—are erasers that reverse acetylation to form closed chromatin with decreased gene expression. In melanoma, HDAC activity is often upregulated, silencing tumor suppressor genes and activating the Mitogen-Activated Protein Kinase (MAPK) pathway. While HDACs silence tumor suppressors, their interaction with the MAPK pathway is bidirectional, involving feedback loops and resistance mechanisms rather than unidirectional activation. Immune evasion with HDAC upregulation has multiple pathways. Histone deacetylation mediated by HDACs suppresses the expression of Major Histocompatibility Complex (MHC) class I molecules and essential components of the antigen processing machinery, such as the proteasome subunits low molecular mass polypeptide (LMP)-2 and LMP-7 and the Transporter Associated with Antigen Processing (TAP) transporter. This suppression occurs through the formation of a more condensed chromatin structure, which inhibits transcription of these genes in melanoma and various other cancer types [99]. Treatment with HDAC inhibitors increases the expression of TAP1, TAP2, LMP2, LMP7, tapasin, and MHC class I molecules in melanoma cells. This upregulation leads to enhanced cell surface expression of class I molecules and costimulatory molecules CD40 and CD86, thereby promoting direct presentation of whole protein antigens and MHC class I-restricted peptides [100]. HDAC2 is recruited to the PD-L1 promoter by STAT1 and facilitates PD-L1 induction by increasing phosphorylation of Janus kinase (JAK)1, JAK2, and Signal Transducer and Activator of Transcription (STAT)1. This process also enhances STAT1 nuclear translocation and its recruitment to the PD-L1 promoter. Knockout of HDAC2 impairs IFN-γ-induced upregulation of H3K27 and H3K9 acetylation, as well as BRD4 recruitment at the PD-L1 promoter [101]. HDAC upregulation is also associated with metastasis. SNAIL directly interacts with the E-cadherin promoter and recruits HDAC1, HDAC2, and the co-repressor Sin3A to the CDH1 promoter to silence E-cadherin expression by deacetylation of histones H3 and H4, an effect that was abolished by HDAC inhibitor trichostatin A treatment [102]. The recruitment of HDACs to the CDH1 promoter is regulated by transcription factor Zinc Finger E-box-Binding Homeobox (ZEB)1, with the Snail/HDAC1/HDAC2 complex essential for enhancer of zeste homolog 2 (EZH2)-mediated repression of CDH1 [103]. HDAC10 suppresses expression of matrix metalloproteinases (MMP)2 and MMP9, genes critical for cancer cell invasion and metastasis, while HDAC11 inhibits migration and invasion of cancer cells by downregulating MMP3 expression [104]. HDAC8 activation in melanocytes and melanoma cells is triggered by various stresses, prompting the cells to adopt a neural crest-stem cell-like state characterized by increased invasiveness and a higher tendency to metastasize to the brain. HDAC8 accomplishes this by deacetylating and inactivating the enzyme EP300. This change enhances EP300’s interaction with Jun-driven genes, while reducing its activity at MITF-controlled genes. As a result, inhibiting EP300 further promotes melanoma cell invasion, stress resistance, and brain metastasis. Overall, HDAC8’s suppression of EP300 shifts gene expression patterns to favor melanoma progression and brain metastasis [105].
1.2.2 Histone Methylation and Demethylation
Histone Methyltransferases (HMTs): writers
Histone methylation activates or represses gene expression associated with melanoma progression. Commonly occurring at lysine or arginine residues, histone methylation determines gene expression based on the site and number of methyl groups added [106,107]. One significant histone lysine methyltransferase (HKMTase, HMT) is the EZH2, the primary component of the polycomb-repressive complex 2 (PRC2). PRC2 induces the trimethylation of histone H3 at lysine 27 (H3K27me3), a mark of tumor suppressor gene silencing. In melanoma, overexpression of EZH2 has been found to cause a high proliferation rate and is associated with aggressive tumor subgroups. There is also accumulating evidence that EZH2 plays a role in the progression and metastasis of melanoma [108,109]. EZH2-mediated H3K27me3 at MHC class I antigen processing pathway (MHC-APP) loci reduces basal expression of these genes and inhibits their interferon (IFN)-γ-induced activation, enabling tumor cells to evade immune surveillance by effector T cells [110]. EZH2-mediated H3K27me3 and DNA methylation repress tumor production of Th1-type chemokines Chemokine (C-X-C motif) ligand (CXCL)9 and CXCL10, which are critical for recruiting effector T cells to the tumor microenvironment. EZH2 inhibits CXCL9 transcription by increasing H3K27me3 at its promoter, thereby impeding CD8+ T cell trafficking to immune-desert tumors [111]. Protein Arginine Methyltransferase (PRMT)1-mediated methylation of EZH2 at arginine 342 strengthens EZH2 binding to target gene promoters and increases H3K27me3 levels, which is necessary for EZH2 to promote the epithelial-to-mesenchymal transition (EMT) program and stimulate cancer cell migration [112].
Other abnormal HMT writer modifications, such as up-regulation of SET domain bifurcated 1 (SETDB1), up-regulation of Lysine methyltransferase 2D (KMT2D), and up-regulation of Euchromatic Histone Methyltransferase (EHMT), also play a role in controlling transcription, chromatin structure, cell differentiation, and melanoma progression. SETDB1 is related to H3K9me3, leading to tumor suppressor gene silencing. Further, H3K4me1 causes activation of thrombospondin-1 (THBS1), which accelerates melanoma initiation and is related to metastasis [93,113]. Orouji et al. found that the activation of thrombospondin-1 (THBS1), which is known to enhance invasiveness and metastasis formation in melanoma, is triggered by SETDB1. In addition to increasing H3K9me3 at a global genomic level, SETDB1 also modifies the methylation patterns and affects H3K4me1 levels upstream of the THBS1 gene at a specific site for transcriptional activation. Thus, SETDB1 may influence not only the distribution of H3K9me3 but also impact other epigenetic markers that control gene activation or suppression. Importantly, using a small molecule inhibitor targeting H3K9me-specific histone methyltransferase to inhibit the SETDB1 protein significantly reduced melanoma cell viability [114]. Additionally, SETDB1 facilitates melanoma cells’ evasion of the immune system by epigenetically silencing genes, particularly endogenous retroviruses. Cuellar et al. show that removing SETDB1 in human leukemia cell lines activates these repetitive elements, leading to increased double-stranded RNAs. When SETDB1 is highly expressed in melanoma cells, it reduces the tumor’s capacity to activate an immune response, promoting immune evasion and resistance to immune checkpoint blockade (ICB) therapies [115].
The Disruptor of telomeric silencing 1-like (DOT1L) gene, often deleted or mutated in human melanoma, exhibits specific mutations that reduce its methyltransferase activity, leading to decreased H3K79 methylation. This reduction affects DNA damage repair by impairing the recruitment of the Xeroderma Pigmentosum complementation group C (XPC) protein to sites of damage, a crucial step in nucleotide excision repair. The findings suggest that DOT1L plays a protective role in preventing melanoma development induced by UV radiation [116].
KMT2D is an HMT responsible for adding a methyl group to histone H3 at lysine 4 (H3K4me1), marking enhancer regions. In melanoma, silencing KMT2D leads to the inactivation of a subset of KMT2D-bound enhancers, which results in decreased H3K4me1 and H3K27ac levels. Silencing KMT2D also downregulates genes critical for cell migration, such as MFGE8 and RPL39L. This alteration promotes tumorigenesis by disrupting enhancer activity [117,118].
PRMT1 modifications enhance the expression of PRMT1 and elevate levels of activated leukocyte cell adhesion molecule (ALCAM) through arginine methylation of histones. This process may contribute to the growth and metastasis of melanomas [119]. PRMT5 is emerging as a target for various solid and hematologic cancers. Its overexpression or dysregulation has been detected in multiple cancer forms, including melanoma.
Histone Demethylases (HDMs): erasers
HDMs such as Jumonji AT-rich interactive domain 1B (JARID1B, KDM5B) are erasers, removing methyl groups from histone 3 at the lysine residue position 4 (H3K4), Jumonji domain-containing protein 3 demethylase (JMJD3) at the lysine residue position 27 (H3K27), and Lysine-specific histone demethylase 1A (LSD1, KDM1A) at the lysine residues at positions 4 and 9 (H3K4 and H3K9). JARID1B is a histone demethylase that has a multifaceted role in melanoma. Research indicates that melanoma cells exhibit increased levels of JARID1B, suggesting that this may occur early in the progression of disease and does not correlate with the invasive phase of melanoma [120]. Chauvistré et al. suggest that JARID1B plays a significant role in tumor growth, maintenance, survival, and treatment resistance. Their research indicates that melanoma cells with elevated JARID1B levels form a slow-cycling stem cell-like subpopulation that can enter a reversible “persister” state crucial for continuous tumor growth. Maintaining this slow-cycling state inhibits melanoma growth and cell invasion. However, these melanoma cells can leverage this state to withstand targeted or cytotoxic therapies. The researchers propose that this idea could be applied as a strategy to improve responses in residual disease following advanced cancer treatments [121]. JMJD3 is a histone demethylase associated with the progression and metastasis of melanoma. JMJD3 is involved in the activation of Nuclear Factor (NF)-κB and Bone Morphogenetic Protein (BMP) signaling pathways that facilitate melanoma development as well as alter the melanoma tumor microenvironment by enhancing angiogenesis and recruiting macrophages [122]. KDM1A, also known as LSD1, is a lysine demethylase that plays a role in several cancers—including melanoma—and is being investigated as a potential therapeutic target. KDM1A has been implicated in the development and progression of melanoma [123,124].
1.2.3 Histone Phosphorylation and Ubiquitination
Histone phosphorylation is implicated in the development of melanoma. It can alter chromatin architecture and influence transcriptional activation, particularly during cellular division [10,125]. Additionally, the phosphorylation of histones H1, H2B, and H3 significantly affects DNA repair mechanisms and gene regulation [10,126].
Ubiquitination is a form of post-translational modification that tags proteins for degradation or modulation of their functions. Ubiquitination influences various cellular processes, including the cell cycle, apoptosis, and DNA repair. Dysregulation within the Ubiquitin-Proteasome System (UPS) is often observed in melanoma, leading to abnormal degradation or stabilization of proteins that can propel tumor growth. Ubiquitination affects B-Raf proto-oncogene, serine/threonine kinase (BRAF) and Mitogen-activated extracellular signal-regulated kinase (MEK) in the MAPK pathway, enhancing survival signaling by stabilizing AKT [127,128]. Specific E3 ligases, such as MDM2, Itchy protein (ITCH), and RING finger protein (RNF)125, regulate oncogenes or tumor suppressors, thereby influencing melanoma progression [129,130]. Deubiquitinases (DUBs), such as USP9X and USP13, stabilize anti-apoptotic proteins, including Myeloid cell leukemia-1 (MCL-1). Targeting the UPS may aid in sensitizing melanoma cells to existing therapies or overcome resistance. The specificity of ubiquitination makes it an attractive target for the development of targeted drugs in melanoma [131].
1.2.4 Other Histone Modifications
Additional less common post-translational histone modifications identified in cancer research include lactylation, citrullination, crotonylation, succinylation, SUMOylation, propionylation, butyrylation, 2-hydroxyisobutyrylation, 2-hydroxybutyrylation, ADP-ribosylation, butolylation, hydroxylation and formylation [132–134]. Lactylation, crotonylation, butyrylation, succinylation, and 2-hydroxyisobutyrylation are types of acylation that bridge cellular metabolism with chromatin regulation. For instance, higher lactate levels in tumors can increase histone lactylation, which changes gene expression. In melanoma, researchers have begun identifying specific histone lactylation sites, such as H3K18la, which is related to poor prognosis [135]. Sirtuin 5 (SIRT5), an eraser of succinylation, is required for proliferation and survival in melanoma [136]. Modifications such as citrullination, SUMOylation, and glycosylation [93] serve as molecular links between cellular stress, DNA damage, immune signaling, and chromatin remodeling in melanoma. Citrullination is catalyzed by peptidylarginine deiminases (PADs), which convert arginine residues to citrulline in histones, thereby altering chromatin structure. SUMOylation, the addition of small ubiquitin-like modifier (SUMO) proteins to histones, is up-regulated in melanoma and supports tumor growth [137]. These histone modifications represent promising avenues as biomarkers providing insights into cellular metabolic states and new opportunities for combination therapies.
1.3 Non-Coding RNA Regulation in Melanoma
Non-coding RNAs (ncRNAs) consist of microRNAs (miRNAs) that are less than 200 bp, typically 21–25 bp single-stranded RNAs, and long non-coding RNAs (lncRNAs) that are more than 200 bp and may extend to over 100 kb.
LncRNAs are involved in gene regulation in melanoma. For example, the lncRNA Survival Associated Mitochondrial Melanoma Specific Oncogenic Non-coding RNA (SAMMSON) gene is targeted by the transcription factor SOX10, with high expression levels in over 90% of human melanomas. Increasing SAMMSON enhances the clonogenic potential of melanoma cells, while its knockdown significantly reduces cell viability, independent of genetic mutations. Silencing SAMMSON disrupts essential mitochondrial functions, specifically in cancer cells [138]. The lncRNA Metastasis Associated Lung Adenocarcinoma Transcript 1 (MALAT1) gene has been shown to promote melanoma cell migration and invasion, as well as inhibit apoptosis. Homeobox (HOX) transcript antisense intergenic RNA (HOTAIR) is suggested to directly interact with histone-modifying enzymes, which alter chromatin structure in melanoma development. Lymph node metastasis in melanoma showed overexpression of HOTAIR. The lncRNA HOTAIR functions as a molecular sponge for the miR-200 family, resulting in miR-200 downregulation and facilitating cancer progression. This regulatory axis disrupts immune checkpoint regulation by inducing epithelial-to-mesenchymal transition (EMT), a process linked to the development of an immunosuppressive tumor microenvironment and resistance to immune checkpoint inhibitors. Consequently, targeting the HOTAIR-miR-200 interaction represents a promising therapeutic strategy to improve immune checkpoint inhibitor efficacy, which is an important therapeutic aspect in melanoma treatment [139,140].
Additional oncogenic lncRNAs, such as BRAF-activated ncRNA (BANCR) and Antisense Non-coding RNA in the INK4 Locus (ANRIL), are found to inhibit apoptosis, promote invasion, and facilitate metastasis in melanoma [141].
MicroRNAs modulate gene expression of approximately 60% of human genes by binding to the 3′ untranslated regions (3′UTR) or 5′UTR of target mRNAs [142–144]. The miRNAs display a complex function as one miRNA can regulate multiple mRNAs, and multiple miRNAs can target one mRNA [145]. They are essential in governing processes such as cell development, growth, differentiation, and maintenance of homeostasis in both normal and diseased cells [146,147].
Research has elucidated a critical role for tumor cell-secreted exosomes throughout different phases of tumor progression and metastasis. These exosomes carry biomolecules, including proteins, RNA, and lipids, from tumor cells to their surrounding environment [148]. Extracellular RNAs (exRNAs) including miRNAs, lncRNAs, and mRNAs are aberrantly expressed in melanoma. Researchers have identified new metabolic reprogramming pathways and therapeutic targets such as the NEAT1-macrophage axis. ExRNAs contribute to melanoma progression by regulating the expression of target genes and mediating key signaling pathways. Melanoma-derived exRNAs reshape the tumor microenvironment. Exosomes containing miR-155 and miR-210 significantly reprogram the metabolism of fibroblasts, resulting in marked reductions in basal and maximal respiration, as well as ATP production, thereby creating a microenvironment that supports metastasis. While exRNA profiles show promise for real-time treatment adaptation and early recurrence detection, more research is needed before miRNAs, mRNAs, and proteins in extracellular vesicles become reliable cancer biomarkers [149]. Translational research has also identified numerous exosomal miRNAs in melanoma that aid tumor evasion of immune response [150].
Dysregulated miRNAs play a crucial role in the development, progression, and treatment resistance of melanoma [151,152]. The central regulatory centers are the oncogenic microRNA (oncomiR) axis and the tumor suppressor network, and the critical downstream targets are the PTEN, MITF, MAPK, and angiogenesis pathways. Epigenetic silencing of tumor suppressor miRNA through DNA methylation and histone modifications is the drug target, leading to clinical trials combining demethylating agents with targeted therapies and immunotherapy. Fig. 2 illustrates the mechanisms of miRNA involvement in the signaling pathways of melanoma. The early and late stages of the disease, along with tumor heterogeneity, influence the paradoxical, positive, and negative regulatory feedback loops that contribute to the fluid and complex nature of melanoma pathogenesis.
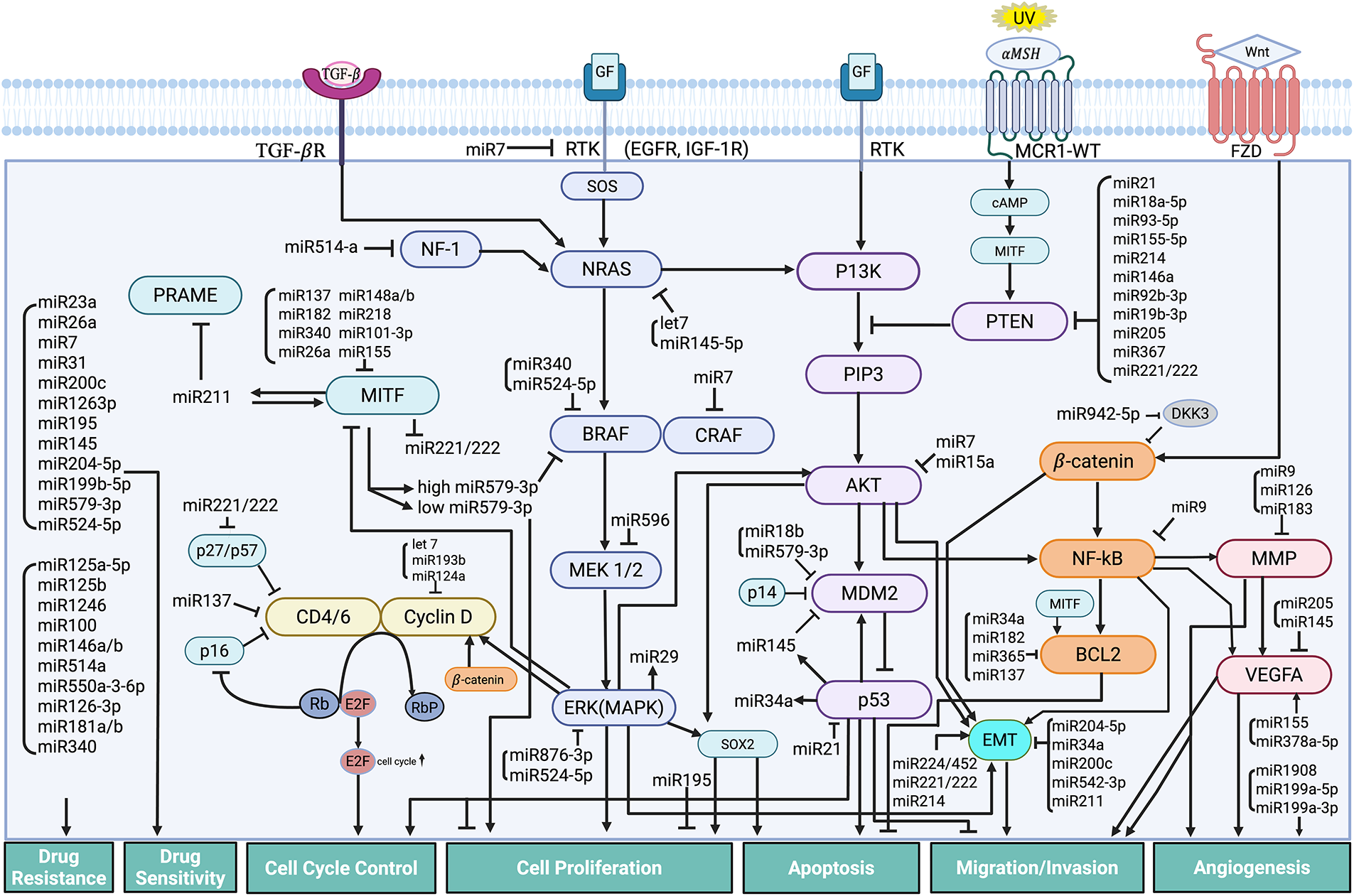
Figure 2: Mechanisms of MicroRNA Regulation in Cellular Pathways of Melanoma. The figure highlights the intricate network of miRNAs in regulating key cellular pathways involved in the development and progression of melanoma. miRNAs modulate multiple signaling cascades that influence critical processes such as cell proliferation, growth, and survival; cell cycle control; cell migration and invasion; apoptosis; drug resistance; and angiogenesis. Abb: PRAME, Preferentially expressed antigen in melanoma; SOX, SRY-box transcription factor; MITF, Microphthalmia-associated transcription factor; CDK, Cyclin-dependent kinase; NF-1, Neurofibromin-1; CDKN2A, Cyclin-dependent kinase inhibitor 2A; NRAS, Neuroblastoma RAS viral oncogene homolog; BRAF, B-Raf proto-oncogene, serine/threonine kinase; CRAF, C-Raf proto-oncogene, serine/threonine kinase; MEK, mitogen-activated extracellular signal-regulated kinase; ERK, Extracellular signal-regulated kinases; PI3K, Phosphoinositide 3-kinase; PIP3, Phosphatidylinositol (3,4,5)-trisphosphate; AKT, Ak strain transforming Protein kinase B; MDM2, Mouse double minute 2 homolog; PTEN, Phosphatase and tensin homolog; GF, Growth factor; ZEB, Zinc enter finger box-binding homeobox; NFkB, Nuclear factor kappa-light-chain-enhancer of activated B cells; BCL2, B-cell CLL/lymphoma 2 protein; EGFR, Epidermal growth factor receptor; IGF, Insulin-growth factor; TGFßR, Transforming Growth Factor beta receptor; FZD, Frizzled receptor; Wnt, Wingless-related integration site; MSH, melanocyte-stimulating hormone; MCR1-WT, melanocortin 1 receptor-wild type; cAMP, cyclic adenosine monophosphate; VEGFA, Vascular endothelial growth factor A; MMP, Matrix metalloproteinase; EMT, epithelial-mesenchymal transition; Rb, retinoplastoma protein; RbP, phosphorylated retinoblastoma protein; E2F, Early 2 Factor. Created in BioRender. Hong, S. (2025)
OncomiRs/Tumor Suppressor miRNAs in Melanoma
Key oncomiRs consistently upregulated include miR-21 [153,154], miR-221/222 [155], miR-214 [156], miR-155 [157], and miR-18a [158] which promote proliferation, invasion, and drug resistance. In contrast, the tumor-suppressing miRNAs that were downregulated consist of miR-200c [159], miR-205 [160], miR-211 [161,162], miR-137 [163], miR-34a/b [164,165], the miR-29 family [166], and the let-7 family [167], which typically inhibit EMT, regulate the cell cycle, and restrain metastasis. miRNA-155 has both oncogenic and tumor suppressive roles.
Specific miRNAs in Melanoma Development
The major specific miRNAs that are crucial for melanoma development are 1) miRNA-21, a master oncomiR targeting multiple tumor suppressors [153,154] 2) miR-221/222 cluster, cell cycle and differentiation regulators [155] 3) miR-29 family, MAPK-responsive tumor suppressors [166] 4) miRNA-34a, p53-regulated master tumor suppressor [164] 5) miRNA-211, paradoxical behavior in melanoma [161,162]; 6) Let-7 family, tumor suppressors targeting RAS and cell cycle genes [167].
miRNA Targets in Signaling Pathways
PTEN-PI3K/AKT axis: Metastasis-associated microRNAs, miR-18a-5p, miR-93-5p, and miR-155-5p, target PTEN collectively, which activates the PI3K/AKT signaling pathway, facilitating melanoma invasion and metastasis [168]. OncomiRs such as miR-21 directly target PTEN, activating the AKT pathway and reducing regulatory T cell (Treg) proliferation [152]. Tumor-secreted miR-214 stimulates Tregs to produce IL-10 by lowering the levels of PTEN. This leads to immune suppression and accelerated tumor growth. Blocking miR-214 has been shown to inhibit Treg activity and slow tumor progression [169]. Up-regulated miR-146a in melanoma accelerates tumor cell growth by activating the NOTCH/PTEN/AKT pathway [170]. The loss of PTEN due to miRNA dysregulation also contributes to resistance to anti-PD-1 immunotherapy by maintaining PI3K/AKT pathway activation, suggesting that a combined approach targeting both PI3K and the miRNA-PTEN interaction may overcome therapeutic resistance in melanoma [171]. Interestingly, miR-92b-3p is the most abundant miRNA in melanoma-derived extracellular vesicles and directly inhibits PTEN, thereby promoting the formation of cancer-associated fibroblasts [172].
MITF-miRNA regulatory network: Tumor-suppressing miRNAs such as miR-148a/b [173], miR-137 [174], and miR-101-3p [175] inhibit melanoma progression by directly targeting MITF. Conversely, MITF influences specific miRNAs, such as miR-211 [161] and miR-579-3p [176], creating feedback mechanisms that govern the shift between proliferative and invasive traits. These MITF-miRNA circuits are increasingly recognized as mediators of BRAF/MEK inhibitor resistance, with the MITF-miR-579-3p axis emerging as both a mechanistic driver and predictive biomarker [176]. An interesting data-driven network approach revealed that specific miRNAs-31, 107, and 222-significantly influence melanoma metastasis and invasion, both individually and in combination, by modulating SOX10, MITF, and their shared targets through various direct and indirect interactions [177].
MAPK pathway modulation: Several miRNAs have been identified that either promote resistance (miR-514a, miR-1246, miR125b) or restore sensitivity to BRAF and MEK inhibitors (miR-7, miR-200c, miR-524-5p, miR-579-3p) by regulating genes involved in autophagy or the RAS/MEK/ERK pathway [178–180]. The oncosuppressor miR-579-3p and the MITF positive feedback regulatory loops govern the balance between proliferation, senescence, and therapeutic resistance in BRAF-mutant melanomas [176]. miR-579-3p and miR-1246 represent promising therapeutic targets and biomarkers in BRAF-mutant melanoma due to their intersection with the MAPK pathway and roles in drug resistance through processes of autophagy and immune checkpoint regulation [181].
p53 pathway and MDM2: The p53-MDM2-miRNA regulatory network is essential for melanoma progression and the development of therapy resistance. Several oncosuppressive miRNAs, such as miR-579-3p, miR-145, miR-23a, and miR-34a, either target MDM2 or act as p53 transcriptional targets, thereby influencing tumor suppressor functions [182,183]. Small molecules that hinder the interaction between p53 and MDM2 can reactivate p53 transcriptional activity and increase levels of tumor-suppressive miRNAs, such as miR-145 and miR-23a [183]. MDM2 inhibitors show clinical promise for restoring p53 function [184] with emerging data supporting synergy with immunotherapy [185].
Wnt/β-catenin signaling: miR-137 functions as a tumor suppressor in uveal melanoma by targeting EZH2, which leads to the suppression of Wnt/β-catenin signaling and EMT [163]. In contrast, miR-942-5p acts as an oncomiR by directly inhibiting Dickkopf-3 (DKK-3), a suppressor of the Wnt pathway [186]. This causes increased nuclear accumulation of β-catenin, thereby aiding in melanoma cell proliferation and invasion.
Angiogenesis pathway: The concept of “angiogenic switch” describes the transition in the tumor microenvironment from a dormant, avascular state to an active state characterized by an increase of pro-angiogenic factors and a decrease of anti-angiogenic factors [187]. Several miRNAs influence endothelial cell activity, vascular endothelial growth factor (VEGF) signaling, and the tumor microenvironment. miRNA dysregulation may facilitate cell invasion and migration in vitro and promote the formation of new vasculogenic structures by melanoma cells, so-called vasculogenic mimicry, presumed to be the result of differentiation plasticity inherent to more primitive, melanoma stem-like cells [188]. miR-378a-5p increases VEGF levels and enhances both in vitro and in vivo angiogenesis [189]. miR-155, derived from melanoma exosomes, promotes the secretion of VEGFA and FGF2, and proteolytic enzymes [148,190].
Epigenetic Mechanisms Silence Tumor Suppressor miRNAs
DNMT1 silencing of miR-211 [191] and combined DNA methylation/H3K27me3 repressing of miR-34a are emerging melanoma-specific targets. Clinically available agents—decitabine, azacitidine, and HDAC/EZH2 inhibitors—offer therapeutic opportunities to restore tumor suppressor networks [192].
miRNA and Membrane Pumps
miRNAs may modulate the expression of membrane pumps responsible for drug efflux directly. An example of this involves the observation that diminution of miR-340-5p levels is related to increased expression of the multi-drug resistance transporter, ATP-binding cassette, sub-family B member 5 (ABCB5), in melanoma cells under oxygen-deprived conditions [193]. Because ABCB5 is expressed by melanoma stem cells [194] and its function also drives pathways that enhance tumor virulence [195], this effect has pleiotropic implications that impact on both therapy resistance and intrinsic tumor aggressiveness.
1.4 Chromatin Remodeling in Melanoma
1.4.1 SWI/SNF Complexes: Structure and Function
SWItch/Sucrose Non-Fermentable (SWI/SNF) complexes are ATP-dependent chromatin remodelers that play a significant role in gene expression by modifying nucleosome positioning. They are essential for processes such as transcription, DNA replication, and repair. These complexes contain a central catalytic subunit, which can either be SWIF/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a (SMARCA)4/Brahma-related gene 1 (BRG1) or SMARCA2/Biologic Response Modifier (BRM), along with various subunits. The complexes include canonical BRG1/BRM-associated factor (cBAF), polybromo-associated BAF (PBAF), and noncanonical BAF (ncBAF) [196,197]. SWI/SNF complexes are vital for melanocyte development and the cellular response to ultraviolet (UV) radiation [198]. Mutations in the genes that encode SWI/SNF subunits are found in 34% of melanomas, with some acting as tumor suppressors and others promoting oncogenesis.
1.4.2 SMARCA4 Heterogeneity and Therapeutic Sensitivity
The impairment of SMARCA4 function may enhance melanoma cell proliferation, invasiveness, and treatment responses. The SMARCA4 status in melanoma may be heterogeneous, with some tumors showing a loss of SMARCA4 function, while in other cases, high levels of SMARCA4 may promote cancer development. The array of transcription factors found in melanoma could determine whether SMARCA4 acts as a tumor suppressor or an oncogene by influencing its genomic location. The activity of SWI/SNF complexes can affect the sensitivity of melanoma cells to different therapies [199]. In a melanoma model, the somatic loss of function of SMARCA4, with the subunit Bptf, suppressed tumor development and disrupted intersecting gene expression programs crucial for tumor cell growth [200]. Research involving uveal melanoma cell lines has demonstrated a response to SMARCA4/SMARCA2 inhibition through small-molecule inhibitors targeting BRM/BRG1 ATPase activity [201].
1.4.3 Other SWI/SNF Subunits in Melanoma
In melanoma cells, BRAF V600E-mediated apoptosis is dependent upon SMARCB1 [202]. SMARCD1 and SMARCD2 both engage with MITF and could play significant roles in the development of melanocytes and the progression of melanoma [203]. SMARCD3 is associated with reduced survival rates in patients with uveal melanoma [204]. Further, AT-rich interaction domain 1A (ARID1A) is the most mutated SWI/SNF gene in cancer [205]. Melanoma patients whose tumors exhibit elevated levels of ARID1A show a higher rate of clinical response to immune checkpoint inhibitors [206]. ARID1B frequently exhibits loss of chromosomal copies in mucosal melanomas [207]. Mutations in ARID2 are linked to exposure to ultraviolet radiation and are observed during the progression to melanoma in situ [208]. A lack of ARID2 enhances the effectiveness of immune checkpoint inhibitors in melanoma by increasing the production of chemokines and facilitating T-cell infiltration [209]. PHD finger protein 10 (PHF10), a component of the PBAF complex, has been found to be highly expressed in cutaneous melanoma and interacts with MYC to stimulate cell proliferation [210]. Elevated levels of BRD7 correlate with reduced survival rates in melanoma patients [211]. BRD9 plays a complex role in melanoma. BRD9 is overexpressed in melanoma and is found to be inhibited by a small molecule, TP-472 [211]. The Nucleosome Remodeling Factor (NURF) complex, with its subunit Bromodomain PHD finger transcription factor (BPTF), is essential for epigenetic modification associated with progression of melanoma. Melanomas show an overexpression of BPTF, which correlates with an unfavorable prognosis and resistance to BRAF inhibitors. Experimental silencing BPTF in melanoma cells resulted in a 65.5% reduction in the proliferative capacity and a 66.4% decrease in the metastatic potential [212]. The loss of Alpha-thalassemia/mental retardation, X-linked (ATRX) has been linked to the progression of melanoma, with decreased levels of ATRX mRNA noted in metastatic cases [213]. In mucosal melanoma, ATRX loss or reduced expression is associated with tumor progression and the alternative lengthening of telomeres (ALT) pathway, while ATRX alterations are less frequent in cutaneous melanoma. An analysis of 21 melanoma cases found that ATRX mutations were connected to loss of ATRX protein expression and ALT, suggesting that ATRX alterations may represent an initial event in conjunctival melanoma development [214]. Furthermore, both ATRX loss and TERT promoter mutations are present in premalignant conjunctival melanocytic lesions, with a majority of metastatic cases exhibiting one of these changes [215].
1.5 RNA Modification in Melanoma
1.5.1 Overview of RNA Modifications
Various RNAs can undergo post-transcriptional modifications that influence cellular stability, localization, and function [216]. Transfer RNAs (tRNAs) exhibit a wide range of modifications [217], while ribosomal RNAs (rRNAs) and non-coding RNA also show a variety of post-transcriptional modifications [218]. Messenger RNAs (mRNAs), similarly, have different types of internal modifications [219]. In melanoma, specific RNA modifications have been linked to tumor development and progression, including N6-methyladenosine (m6A) and adenosine-to-inosine (A-to-I) editing. The upregulation of m6A modification regulatory proteins and A-to-I editing alter the sequences of proteins.
1.5.2 m6A Modification Machinery: Writers, Erasers, Readers
Modification of m6A is a reversible and tightly regulated process involving three types of enzymes and binding proteins—writers, erasers, and readers—that collaboratively control the specific placement of m6A on RNA. This process governs RNA functions such as nuclear transcription, export, cytoplasmic stability, translation, and spatial regulation [220]. Writers, such as Methyltransferase-like 3 (METTL3), METTL4, and RBM15/15B, serve as methyltransferases and form complexes that install m6A marks in the nucleus. Erasers, including AlkB homolog 5 (ALKBH5) and Fat mass and obesity-associated (FTO), act as demethyltransferases, removing these methyl groups to fine-tune their distribution [221]. Reader proteins, namely YT521-B homology domain (YTHD)C1/2 in the nucleus and YTHDF1/2/3 in the cytoplasm, interpret these marks to influence RNA splicing, transport, stability, and translation. Through their coordinated activity, these writer, eraser, and reader enzymes govern various aspects of RNA metabolism [222].
1.5.3 m6A Modifications and Melanoma Progression
Wang et al. performed single-cell and spatial RNA-seq in T cells, which revealed that elevated m6A modification contributes to melanoma virulence and is associated with decreased immune cell infiltration; the expression of m6A readers (YTHDF2, RBM15B) and writers (METTL3, ZC3H13) was reduced, while the m6A eraser (FTO) was upregulated [223].
A study examining the epitranscriptomic profile of melanoma through several publicly accessible databases highlighted DNMT3A and METTL4 as key potential regulators of melanoma growth. Functional validation of DNMT3A and METTL4 was performed using shRNA-mediated knockdown, demonstrating that their depletion in melanoma cells resulted in inhibited cell growth [224].
A study showed that METTL3-mediated m6A hypermethylation of uridine-cytidine kinase 2 (UCK2) enhanced melanoma cell metastasis by activating the Wingless-related integration site (WNT)/β-catenin signaling pathway [225]; UCK2 is the key rate-limiting enzyme in the pyrimidine nucleotide salvage pathway, thus playing a significant role in promoting cancer cell invasion, proliferation, and metastasis [226]. Additionally, METTL3 promotes EMT and thereby promotes cancer cell migration and invasion [227]. Melanoma cells in particular exhibit elevated METTL3 expression compared to normal melanocytes, indicating that METTL3 may play a role in regulating proliferation, invasion, migration, and drug resistance of melanoma cells [228].
The mutation and expression levels of the ALKBH5 gene in melanoma patients are associated with their response to immunotherapy. Furthermore, a small-molecule inhibitor of ALKBH5 has been shown to improve the effectiveness of cancer immunotherapy [229]. ALKBH5 enhances the stability and expression of FOXM1 mRNA by removing m6A methylation, thereby inducing epithelial-mesenchymal transition (EMT) and promoting melanoma metastasis [230].
Research has shown that when FTO demethylates mRNA, it can stimulate the growth of melanoma and diminish the effectiveness of anti-PD-1 immunotherapy. When FTO was knocked down in mice, melanoma cells exhibited increased sensitivity to interferon-gamma and showed improved responses to anti-PD-1 therapy [231].
1.5.4 A-to-I RNA Editing in Melanoma
A-to-I editing has been implicated in both the promotion and inhibition of tumor growth in melanoma. The editing of miR-455-5p by Adenosine Deaminase Acting on RNA 1 (ADAR1) may enhance metastasis, while editing of miR-378a-3p can suppress metastasis of melanoma. Alterations in A-to-I editing levels in melanoma cells have also been associated with resistance to apoptosis. Additionally, A-to-I editing can aid in immune evasion by altering the immunogenic properties of specific RNA structures, which hinders recognition by the immune system [232]. Furthermore, RNA editing signatures have been found within genes that can differentiate patients’ responses to immunotherapy among various patient groups [233].
1.5.5 Future Directions Utilizing RNA Modification
Preclinical studies have highlighted the significant roles of both m6A and adenosine-to-inosine (A-to-I) RNA editing in various diseases, including cancer, chronic inflammation, and infections, fueling interest in their therapeutic applications. Structural insights into ADARs and key m6A enzymes have enabled the development of small-molecule inhibitors that modulate their expression, catalytic activity, or RNA binding. Additionally, targeting RNA modifications and their regulators may enhance the effectiveness of immunotherapies by modulating the tumor immune microenvironment [234].
2 Epigenetic Biomarkers in Diagnosis and Prognosis of Melanoma
2.1 Methylation-Based Biomarkers
2.1.1 MGMT Promoter Methylation as Biomarkers
The most extensively studied epigenetic modifications in melanoma involve DNA methylation patterns, with multiple markers demonstrating diagnostic and prognostic value across independent cohorts [69]. Among all epigenetic markers, only MGMT promoter methylation currently plays a role in guiding therapeutic decisions for melanoma, although its adoption varies substantially among institutions [57]. Routine MGMT testing in melanoma centers remains limited, primarily because immunotherapy and targeted therapy have largely replaced chemotherapy as first-line treatments. Epigenetic silencing of MGMT may sensitize melanoma cells to alkylating agents such as temozolomide by impairing their ability to repair chemotherapy-induced DNA damage [235]. One study of patients with stage IV cutaneous melanoma showed that MGMT promoter methylation is associated with a significantly improved response rate to the single-agent dacarbazine/temozolomide [80]. The DeCOG study found no significant association between MGMT promoter methylation status and treatment response, progression-free survival, or overall survival in melanoma [236]. MGMT promoter methylation may help identify melanoma patients who would benefit from melphalan regional chemotherapy [237].
2.1.2 RASSF1A Hypermethylation as Biomarkers
RASSF1A hypermethylation serves as a diagnostic and prognostic marker in melanoma. Melanoma cell lines show 69% methylation [238], and patient tumors demonstrate a striking stage-dependent pattern, occurring in 0% of stage I/II primary melanomas increasing to 26.7%–27.8% in stage III disease and 48.9% in stage IV metastatic melanoma [71,72,239]. RASSF1A methylation exhibits limited independent prognostic significance, as evidenced by TCGA analysis of 355 patients, which revealed no significant association with disease-free survival (HR = 0.94, p = 0.694) or overall survival (HR = 0.74, p = 0.106). However, it may predict a poor biochemotherapy response when highly methylated [240]. Methylation of the RASSF 1 A promoter in cell-free DNA is detected more frequently in melanoma patients than in healthy controls. Evaluation of this marker in plasma revealed high diagnostic accuracy for melanoma, with an area under the curve of 0.905. Although there is no significant correlation between cell-free DNA methylation and circulating tumor cells individually, combining both biomarkers improves the detection rate for invasive and metastatic melanoma [241].
2.1.3 CDKN2A Promoter Methylation as Biomarkers
Approximately 25% of cutaneous melanoma metastases show hypermethylation of the CDKN2A promoter, with p16(INK4A) promoter methylation specifically detected in 15 of 59 metastatic cases. This alteration is significantly more common in NRAS-mutated tumors than in those without NRAS mutations (p = 0.0004), suggesting a link between NRAS mutations and p16(INK4A) methylation [242]. Methylation-induced loss of p16 function is an independent predictor of poor survival (HR 2.5) [243] and is closely associated with more aggressive clinical features, NRAS mutations, and the transition from primary to metastatic melanoma.
2.1.4 CpG Island Methylator Phenotype (CIMP) as Biomarkers
The CIMP identifies melanoma patients with an increased risk of death, with a hazard ratio of 11.84 (95% CI: 4.65–30.20) compared to tumors with low methylation. CIMP-positive melanomas are more frequently observed in patients aged 65 or older, are associated with lentigo maligna melanoma histology, ulceration, advanced AJCC stage, and a lower number of tumor-infiltrating lymphocytes [244]. The CIMP gene panel consists of Methylated-IN-Tumour (MINT)17, MINT31, Tissue factor pathway inhibitor 2 (TFPI2), Wnt inhibitory factor 1 (WIF1), RASSF1A, and Suppressor of cytokine signaling 1 (SOCS1) [239] as well as further hypermethylation observed in PTEN, Vitamin D Receptor (VDR), PD-L1, and TET2 [244]. Importantly, CIMP may be detectable as early as stage 1b, indicating its potential value for early risk assessment, if confirmed in future studies. Fig. 3 illustrates a forest plot of the meta-analysis of various DNA methylation biomarkers that are relevant to melanoma prognosis. Most of these biomarkers indicate an increased risk of mortality (Hazard Ratio [HR] > 1).
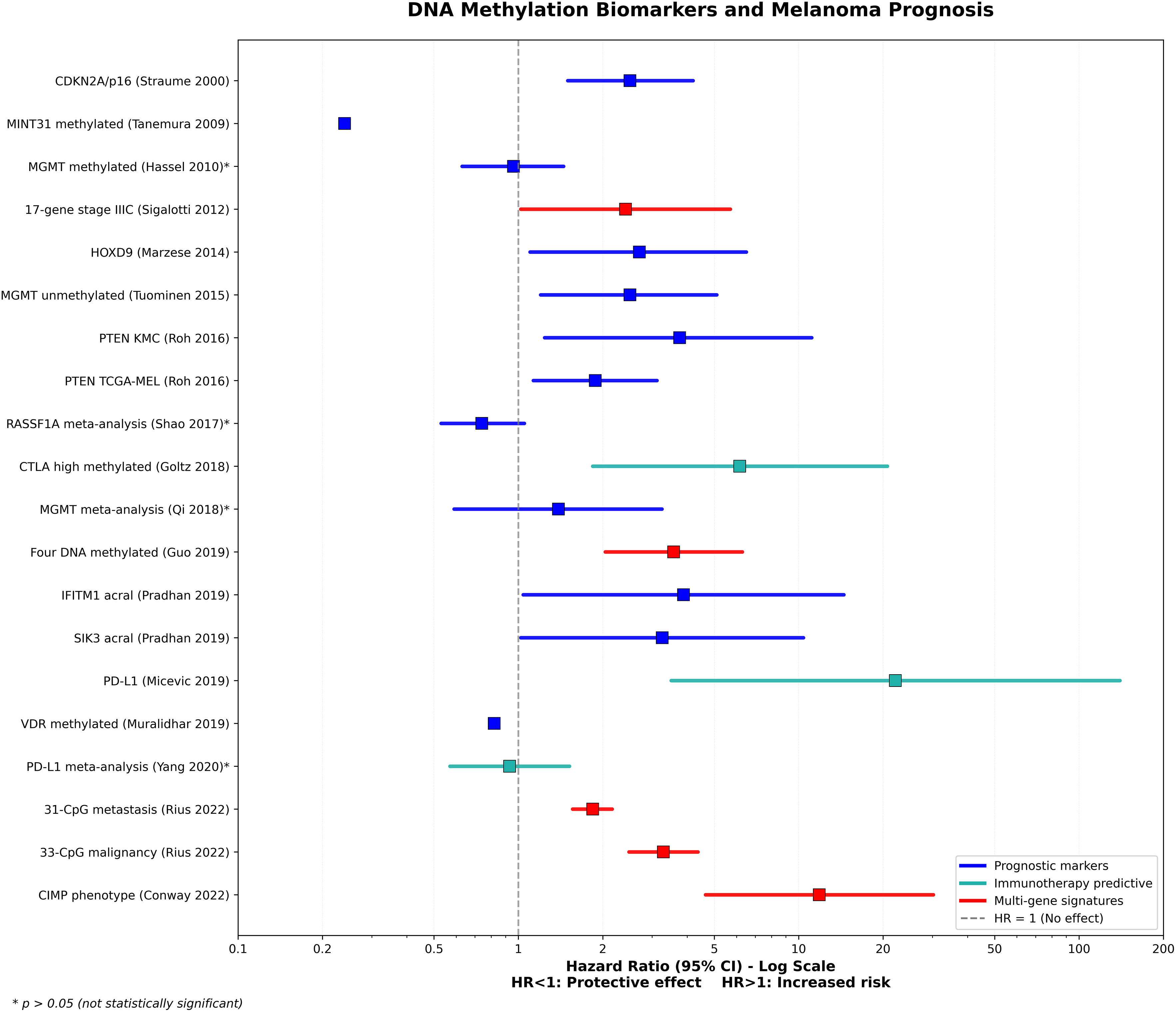
Figure 3: DNA methylation biomarkers and melanoma prognosis. Most DNA methylation biomarkers demonstrate increased mortality risk (Hazard Ratio [HR] > 1), with immunotherapy-predictive markers showing particularly strong associations—Programmed death-ligand 1 (PD-L1) [245] expression (HR = 22.1) and Cytotoxic TLymphocyte-Associated Protein 4 (CTLA-4) [246] high methylation (HR = 6.16). A multi-gene signature, CpG Island Methylator Phenotype (CIMP) [244] phenotype, indicates an elevated risk (HR = 11.84). Among individual prognostic markers, Phosphatase and TENsin homolog (PTEN) [247] and homeobox D9 (HOXD9) [248] show moderate risk associations, while Methylated-IN-Tumour (MINT)31 [239] and Vitamin D Receptor (VDR) [249] methylation demonstrate protective effects (HR < 1) [80,236,240,243,250–255]. CI: Confidence Interval
2.1.5 5-hmC as a Diagnostic Biomarker
Identifying dependable ancillary markers to assist in the classification of melanocytic lesions continues to be a significant challenge. One of the most promising and well-documented epigenetic biomarker candidates is 5-hmC, an oxidized form of 5-methylcytosine produced by the TET family of dioxygenases. In benign melanocytic nevi, nuclear 5-hmC is expressed in high levels, but there is a progressive decrease in dysplastic nevi, Spitz tumors, atypical borderline melanocytic lesions, and invasive melanomas [44,58,256,257]. This gradual loss has been documented in various independent studies, suggesting that 5-hmC serves as an epigenetic marker for malignant transformation in melanocytes.
Extensive retrospective research has validated the diagnostic utility of 5-hmC immunohistochemistry. Rodic et al. found that employing a semi-quantitative 5-hmC scoring system could differentiate between nevi and melanomas, achieving over 90% sensitivity and specificity [256]. Further investigations by Yu and colleagues corroborated these results, indicating that 5-hmC and PRAME exhibit complementary expression patterns, with the loss of 5-hmC combined with diffuse PRAME positivity being particularly indicative of melanoma [258]. In their study of 144 lesions, the area under the ROC curve (AUC) was 0.91 for 5-hmC alone and improved to 0.97 when paired with PRAME, emphasizing the advantage of using these markers together.
Despite its diagnostic value, interpreting 5-hmC results does come with challenges. 5-hmC is a fundamental epigenetic regulatory modification that is ubiquitously present across cell types and is not lineage-specific. This often necessitates double labeling by immunohistochemistry (IHC) with melanocytic markers, such as Melanoma Antigen Recognized by T cells (MART)-1, to accurately assess 5-hmC level in the melanocytic lesional cells [259,260]. Moreover, 5-hmC staining typically shows a continuous gradient rather than a clear dichotomy, compromising the reproducibility of semi-quantitative scoring across different observers [47,261]. Additional internal control issues may surface due to 5-hmC regulation in keratinocytes and neighboring epithelial cells, with adjacent squamous dysplasia or actinic damage potentially complicating interpretations [262,263]. These complications might explain the variable levels of 5-hmC in melanocytic neoplasms. In addition, the understanding of mechanisms of loss 5-hmC in melanoma remains incomplete. We observed that specific melanoma subtypes, such as acral or desmoplastic melanoma, do not exhibit significant loss of 5-hmC compared to other melanoma subtypes, and the underlying mechanism remains unclear [264,265].
To address subjective biases, recent studies have utilized computer-assisted digital image analysis. Research of standardized image quantification in dysplastic nevi and superficial spreading melanomas revealed that 5-hmC levels in the epidermal/junctional area offered the most significant differentiation, with ROC-AUC values between 0.76 and 0.79 [266]. This work highlights the potential of digital pathology, supported by AI-assisted algorithms, to enhance reproducibility and enable more precise reporting frameworks. Similar methodological improvements have been investigated in other groups [267,268].
Overall, the evidence strongly endorses 5-hmC as a beneficial addition to the histopathological assessment of melanocytic tumors. Its optimal diagnostic utility lies best in conjunction with other markers such as PRAME [258], p16 [58], and Ki-67 [264]. Fig. 4 illustrates a workflow diagram of the integration of epigenetics into the diagnosis of an ambiguous melanocytic lesion and treatment of high-risk melanoma.
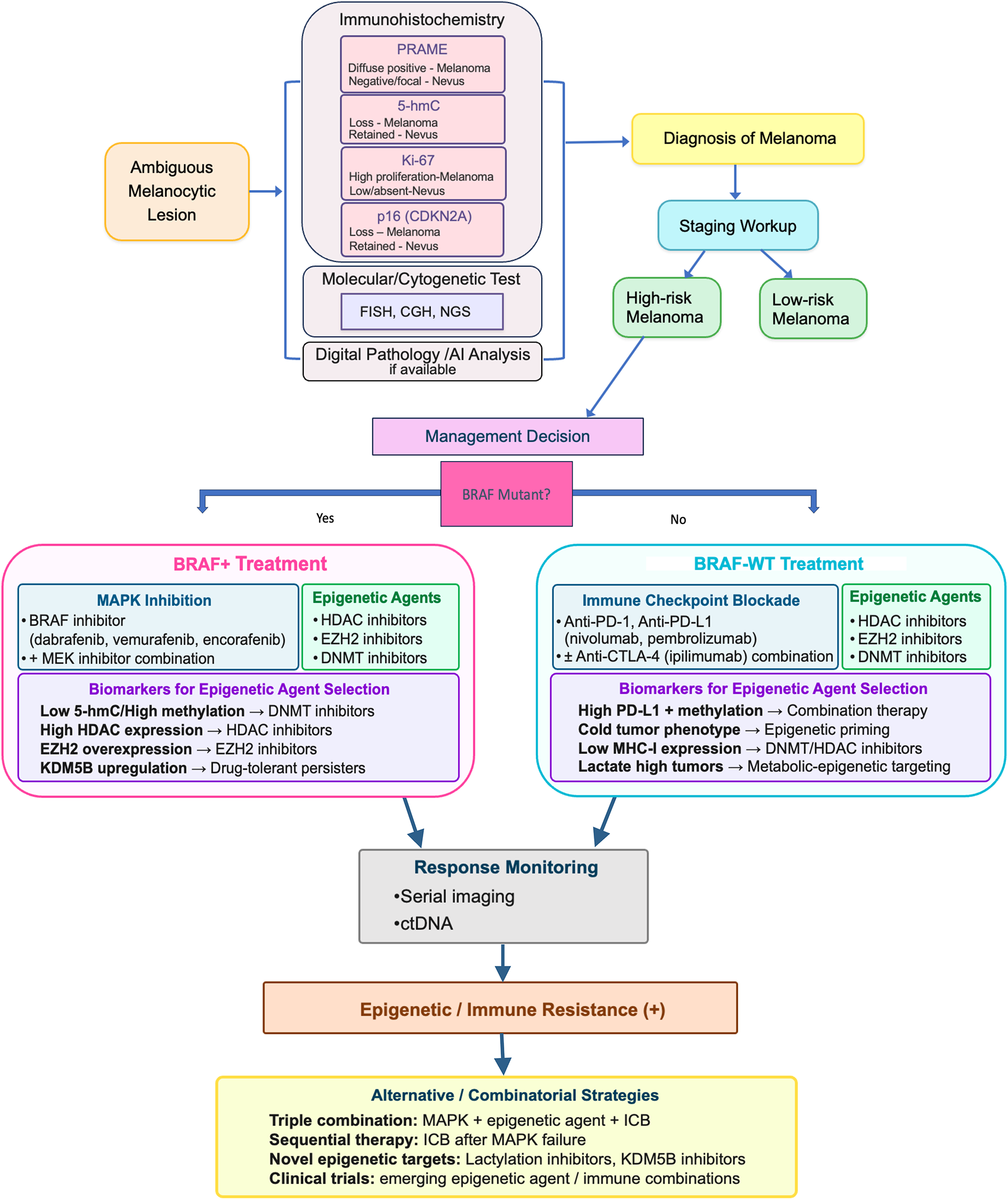
Figure 4: Integration of epigenetics into diagnosis and management of melanoma. An ambiguous melanocytic lesion diagnosis may benefit from epigenetic biomarkers as ancillary tests. Management of high-risk melanoma may include epigenetic agents with clinical trial enrollment, and epigenetic biomarkers for treatment selection. Abb: 5-hmC, 5-hydroxymethylcytosine; PRAME, Preferentially Expressed Antigen in Melanoma; CDKN, Cyclin-Dependent Kinase Inhibitor; FISH, Fluorescence In-Situ Hybridization; CGH, Comparative Genomic Hybridization; NGS, Next-Generation Sequencing; AI, Artificial Intelligence; ctDNA, Circulating tumor DNA; HDAC, Histone Deacetylase; DNMT, DNA Methyltransferase; EZH2, Enhancer of Zeste Homolog2; KDM5B, Lysine Demethylase 5B; MHC, Major Histocompatibility Complex; MAPK, mitogen-activated protein kinase; BRAF-WT, B-Raf proto-oncogene, serine/threonine kinase-wild type; MEK, mitogen-activated extracellular signal-regulated kinase; PD-1, programmed cell death protein-1; PD-L1, programmed death-ligand1; CTLA-4, Cytotoxic T-lymphocyte-associated protein-4; ICB, Immune Checkpoint Blockade
Moreover, 5-hmC has prognostic and mechanistic relevance [263]: its reduction is associated with increased tumor aggressiveness and adverse outcomes, and reinstating 5-hmC via TET/IDH pathways reduces melanoma growth in experimental settings [44,269]. As digital pathology and molecular testing advance, incorporating 5-hmC into multi-parameter panels shows great promise for enhancing diagnostic precision [263], aiding in risk assessment, and potentially guiding therapeutic innovations in melanoma.
2.2 MicroRNA Panels for Early Detection/Relapse Prediction
Recent advancements in miRNA-based liquid biopsy techniques have yielded encouraging results for the early detection and relapse prediction in melanoma [145].
Circulating miRNA biomarkers have demonstrated a sensitivity of 87% and specificity of 81% in various meta-analyses [270]. Several validated panels have been developed, including the MEL38 signature, which consists of 38 circulating plasma miRNAs and has shown strong independent classification accuracy (AUC 0.79–0.94) when analyzed using a support vector machine algorithm [271]. The MEL38 diagnostic signature classifies patients into distinct diagnostic categories using RNA sequencing from either solid tissue or plasma. MEL38 scores were able to differentiate between metastatic and locally invasive melanoma samples, as well as between melanoma in situ and normal skin samples. However, MEL38 scores could not differentiate between benign nevi and melanoma in situ samples [272].
A specific 4-miRNA plasma extracellular vesicle panel has shown to non-invasively diagnose melanoma with an AUC ranging from 0.75 to 1.00 [273]. A meta-analysis and systematic review conducted by Jones and Nonaka evaluated 898 melanoma patients across 9 studies using circulating miRNA panels and reported 89% sensitivity, 85% specificity, a diagnostic odds ratio of 45, and an AUC of 0.93 [274].
Friedman et al. demonstrated that serum miRNAs may be used as biomarkers for both monitoring melanoma tumor burden over time and accurately identifying primary melanoma patients at elevated risk of recurrence [275]. A study by Stark et al. highlighted that the prognostic and predictive capabilities of the MELmiR-7 panel have been shown to predict the overall survival of melanoma patients more effectively than both serum lactate dehydrogenase (LDH) and S100B levels. microRNAs associated with melanoma [276]. Moreover, a combination of four miRNAs (miR-150, miR-30d, miR-15b, and miR-425) was effective in differentiating patients based on recurrence-free and overall survival, improving the prediction of recurrence beyond stage classification. It was observed that serum miR-15b levels increased significantly over time in patients who experienced recurrence, while in non-recurrent patients, these levels did not show significant changes over time. The demonstration that miR-15b rises progressively only in recurrence cases provides biological evidence for a window of opportunity where enhanced surveillance or earlier therapeutic intervention may improve outcomes [277]. In the case of solid tissue, the prognostic MEL12 signature categorizes patients into low-, intermediate-, and high-risk groups, regardless of clinical covariates. The hazard ratios for 10-year overall survival, based on survival intervals, were found to be 2.2 (high-risk vs. low-risk) and 1.8 (intermediate-risk vs. low-risk), surpassing those of other existing prognostic models [272]. Lastly, a study identified specific circulating cell-free miRNAs in plasma samples from melanoma patients with brain metastasis, which may provide insights into assessing those at risk for developing melanoma brain metastasis [278].
2.3 Liquid Biopsy and Circulating Epigenetic Markers
2.3.1 Overview and Current Landscape
The liquid biopsy field is undergoing rapid advancements, with 2024 marking a high point in published research on the subject [279]. Blood remains the primary biofluid studied, with a focus mainly on circulating tumor DNA (ctDNA) as the most extensively analyzed component. Other significant components include circulating tumor cells (CTCs), nucleosomes, lncRNAs, and miRNAs, which are found in exosomal vesicles or as cell-free RNA [280].
2.3.2 ctDNA vs. miRNA: Comparative Advantages
Unlike ctDNA, which is released from necrotic or apoptotic tumor cells and consequently may lead to low and inconsistent levels in circulation, circulating miRNA can be released by cancer cells regardless of their viability. ctDNA has a short half-life of less than two hours, which can complicate laboratory processes such as sample preparation, storage, and transportation, potentially affecting the accuracy of the results. In contrast, miRNAs are stable in circulation since they are packed in exosomes or encapsulated lipid particles and may also be protected by nucleophosmin, microvesicles, and high-density lipoproteins [145].
2.3.3 Advantages and Limitations of Epigenetic Liquid Biopsy
The advantages of utilizing epigenetic modifications in liquid biopsies include their ability to provide early insights into tumor development, details about tissue origin, diverse features, and the potential for reversibility and dynamism. The disadvantages include limitations due to low quantities and a lack of standardized detection and analysis methods [281]. Research on epigenetic liquid biopsy in melanoma is not as extensive as in other cancers, such as breast, colorectal, lung, and ovarian cancers. Other cancer research has yielded promising findings regarding ctDNA of 5-mC and 5-hmC, as well as nucleosome and histone post-translational modifications, ncRNAs, and miRNAs. Multiple epigenetic markers are often combined for clinical application [281].
2.3.4 ctDNA as a Prognostic Marker in Melanoma
The prospective PET/LIT study enrolled 104 patients and demonstrated that ctDNA has a prognostic value in patients with advanced melanoma treated with immune checkpoint inhibitors. Using patient-specific mutations, tumor-informed panels are designed in 1 week, and ctDNA can be sequenced and analyzed in 2 weeks, enabling personalized monitoring. Although the cohort size was small, ctDNA was detected in 76.9% of adjuvant patients who experienced relapse, whereas all patients without disease progression remained ctDNA-negative [282]. In a prospective clinical trial of ctDNA for advanced melanoma, post-surgery positive ctDNA was associated with worse overall survival (HR 6.04) and systemic relapse-free survival (HR 3.77), with ctDNA detection preceding radiological relapse by a median of 4 months [283].
2.3.5 Emerging Biosensor Technologies
New advances in liquid biopsy introduce biosensor technologies enabling point-of-care testing with nanomolar detection limits. Optical biosensors offer a significant opportunity in liquid biopsy due to their enhanced sensing performance and practical, user-friendly properties. The technology has demonstrated sensitive detection of proteins, peptides, ctDNA, miRNA, exosomes, and CTCs [284]. The distinct mutations of ctDNA make them detectable targets via electrochemical methods, and small non-coding RNAs have been the focus of electrochemical detection due to their differential expression in cancer cells [285].
3 Epigenetic Therapy in Melanoma
3.1 Epigenetic Therapeutic Agents
The effectiveness of epigenetic drugs as single agents in solid tumors remains limited, unlike their significant clinical efficacy in certain hematological malignancies [286]. The clinical trials have revealed an unexpectedly high incidence of side effects that need further scrutiny. The major epigenetic drugs that are in clinical trials are DNMT inhibitors, HDAC inhibitors, and EZH2 inhibitors (Fig. 4). Table A2 lists the recent clinical trials of epigenetic agents for melanoma. HDAC inhibitors are the majority of all the phase I, I/II, and II trials, followed by DNMT1 inhibitors (Fig. 5). Currently, there are no epigenetic drugs that have received FDA approval specifically for the treatment of melanoma.
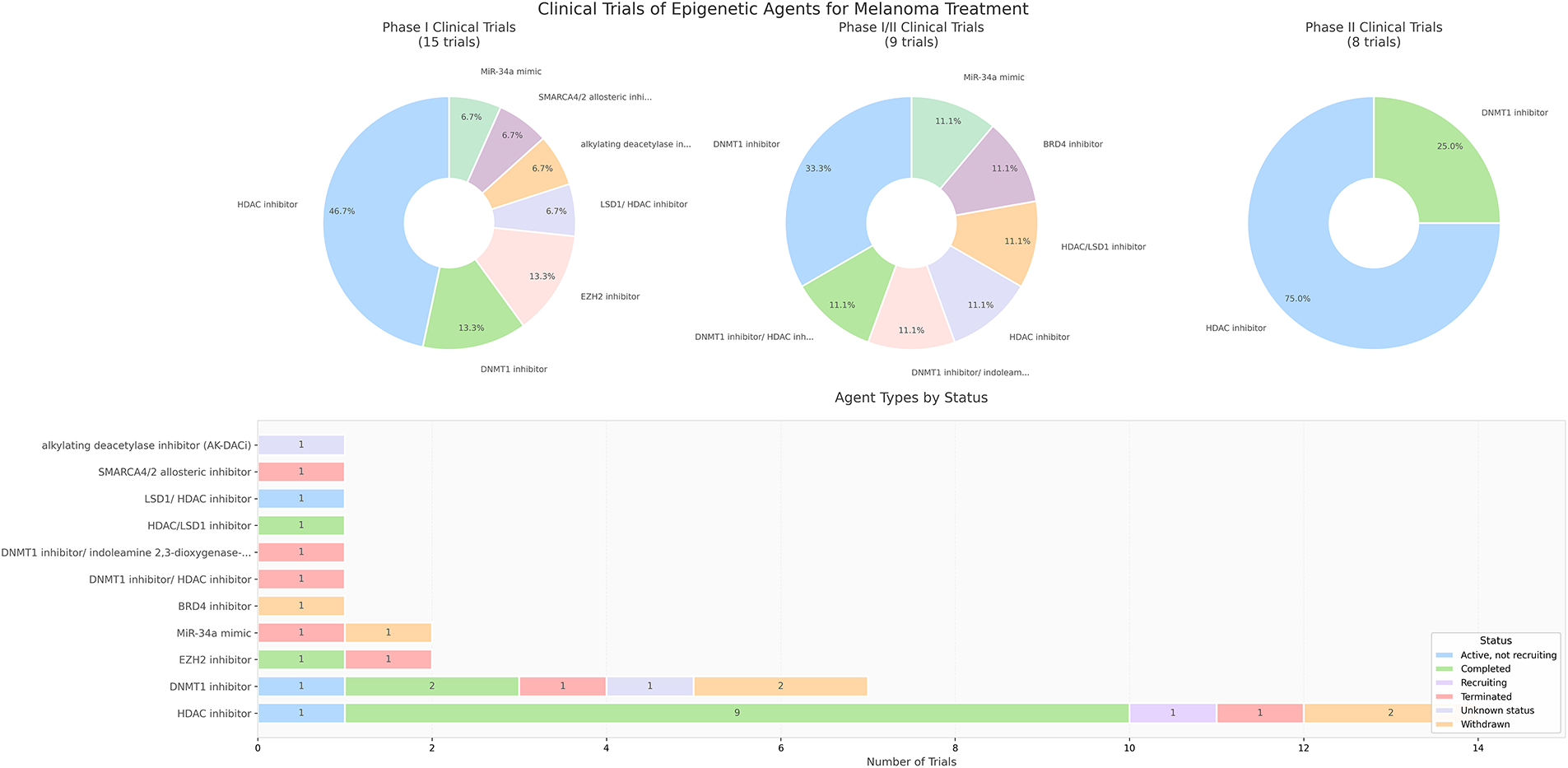
Figure 5: Distribution of current clinical trials of epigenetic agents for melanoma treatment. Phase graphs show the distribution of agents in each phase, and the bar graph shows the trial status of each epigenetic agent
DNMT inhibitors are designed to inhibit the activity of DNA methyltransferases, leading to the reactivation of silenced tumor suppressor genes. Preclinical studies have shown that these inhibitors can re-sensitize melanoma cells to conventional chemotherapy and targeted therapies. However, clinical trials of these agents in melanoma have shown mixed results, highlighting the need for more specific and effective agents [192]. HDAC inhibitors (HDACi) are among the most well-explored epigenetic drugs for melanoma treatment. These inhibitors can induce histone hyperacetylation, leading to chromatin relaxation and reactivation of tumor suppressor genes.
HMT inhibitors, such as EZH2 inhibitors, are emerging as a potential therapeutic strategy. By inhibiting EZH2, these drugs aim to reactivate the expression of tumor suppressor genes, thereby slowing melanoma progression and metastasis. Early phase clinical trials are investigating the safety and efficacy of these inhibitors in combination with other treatments [192]. JQ1, an inhibitor of BRD4, shows promise in enhancing immunotherapy for melanoma by modulating immune responses and inhibiting tumor progression. It can suppress the expression of c-MYC and PD-L1, which may lessen tumor-induced immunosuppression and enhance the effectiveness of treatments such as PD-1/PD-L1 blockade [287]. JQ1 has also been investigated to enhance the apoptosis of B16 melanoma cells by modifying mitochondrial dynamics, which leads to mitochondrial dysfunction and an elevation in oxidative stress [288]. Targeting non-coding RNAs is another exciting avenue for epigenetic therapy in melanoma. miRNA-based therapeutics are being developed to either mimic tumor-suppressing miRNAs or inhibit oncogenic miRNAs [289].
3.2 Combination Strategies with Immunotherapy and Targeted Therapy
3.2.1 Overview of Epigenetic Combination Approaches
Epigenetic agents have been combined with chemotherapies, immunotherapies, and targeted therapies, including other epigenetic drugs, to target parallel tumorigenic pathways, enhance cancer cell death, and overcome resistance mechanisms. Despite these efforts, epigenetic combination therapies have achieved limited success in solid tumors. The underlying cause of poor efficacy may suggest that solid tumors are biologically more complex or are less reliant on epigenetic modifications [286].
Clinical trials investigating HDACi in combination with other melanoma therapies are showing some therapeutic efficacy [192]. While Vorinostat has not received FDA approval for melanoma, patients with BRAFi/MEKi-resistant BRAFV600-mutated melanoma have tolerated intermittent treatment with vorinostat well, and 9% of these patients experienced lasting antitumor responses [290].
3.2.2 Emerging Epigenetic Targets for Combination Strategies
Novel epigenetic treatment such as targeting PRMT1 may be considered for synergistic combination therapy with checkpoint inhibitors, as PRMT1 has shown to enhance interferon signaling by increasing the levels of endogenous retroviral elements [291]. Another new insight demonstrates how understanding epigenetic resistance mechanisms can lead to effective, low-toxicity therapeutic strategies using FDA-approved drugs, such as statins, and suggests that Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1α alpha) [292] status could be routinely included in molecular profiling to guide treatment decisions [293].
3.2.3 Clinical Trials for Checkpoint Inhibitor-Refractory Patients
For patients with cutaneous melanoma who have progressed or are refractory to checkpoint inhibitor therapy, class I HDAC inhibitor (entinostat or domatinostat) + anti-PD-1 rechallenge has been tried. The SENSITIZE trial with Domatinostat + Pembrolizumab objective response rate (ORR) is 7.5% and disease control rate (DCR) is 30% (NCT03278665. Table A2). The ENCORE-601 trial with Entinostat + Pembrolizumab did not achieve the primary response rate endpoint but provided a clinically meaningful benefit, with objective response in 9% of patients. No new toxicities, including immune-related adverse events, were seen for either drug [294]. The rationale centers on the ability of HDAC inhibitors to enhance tumor antigen presentation and reverse immune evasion mechanisms.
3.2.4 Strategies for BRAF/MEK Inhibitor-Resistant Melanoma
For patients with BRAF-mutant melanoma who have developed resistance to BRAF/MEK inhibitor, HDAC inhibitor or DNMTi + continued/rechallenge of BRAF/MEK inhibitors has been tried. The most recent trial regimen is Tazemetostat + BRAF/MEK inhibitors [NCT04557956. Table A2]. For carefully selected treatment-naive patients with high-risk features, investigational approaches have been considered. For low tumor mutational burden (TMB) with hypermethylation signature, DNMTi + immunotherapy has been considered. For “Cold” tumor phenotype, HDACi + immunotherapy has been considered.
3.2.5 Current Status of Epigenetic Agents
Until definitive Phase III validation becomes available, epigenetic agents remain as investigational tools that expand treatment options for patients with limited alternatives.
The integration of epigenetic therapies into melanoma treatment represents an evolving frontier that requires continued research, careful patient selection, and thoughtful clinical implementation within appropriate investigational frameworks.
3.3 Clinical Trial Failure Analysis
3.3.1 Reasons for Clinical Trial Failures
Epigenetic drugs have not received FDA approval for melanoma treatment, despite clear pharmacodynamic activity. The main reasons for failure were the lack of patient selection biomarkers, use of maximum tolerated doses that caused excessive toxicity before reaching optimal biological doses, and the misconception that monotherapy would be effective in a disease that requires combination therapy. Single-agent trials showed low objective response rates. However, when these agents were combined with checkpoint inhibitors, response rates improved and remissions were more durable. These results indicate that strategic combinations guided by predictive biomarkers are a more promising approach than abandoning epigenetic therapies.
3.3.2 Mechanisms of Immune Evasion in Cancer
Recognizing how cancer evades the immune system and discovering the concept of immune priming marked a critical turning point in the development of new treatment strategies. Immune evasion by cancer cell is explained by various mechanisms. Tumors promote an embryonic-like gene expression pattern in endothelial cells through angiogenesis, thereby hindering leukocyte infiltration and weakening antitumor immunity [295]. Immune evasion also starts from abnormal tumor blood vessels reducing blood flow and intensifying existing hypoxia within the tumor [296]. This hypoxic environment not only increases resistance to chemotherapy and radiation but also promotes immune escape by amplifying the activity of regulatory T cells, myeloid-derived suppressor cells, and tumor-associated macrophages [297].
3.3.3 Epigenetic Immune Priming Strategy
Preclinical and early clinical studies have shown that several epigenetic therapies can prime the immune system, both as monotherapies and in combination with immune-based treatments [298]. Furthermore, metronomic chemotherapy has been shown to reduce immunosuppressive cells in the tumor microenvironment, such as regulatory T cells and myeloid-derived suppressor cells. Immune checkpoint inhibitors complement this effect by sustaining T cell activation and boosting anti-tumor responses. When combined, these strategies work synergistically to strengthen anti-tumor immunity [299]. Importantly, low-dose epigenetic priming with DNMTi and HDACi has proven more effective than standard maximum tolerated or prolonged dosing in melanoma, achieving sustained immunomodulation without cytotoxicity [300]. This was demonstrated in the NIBIT-M4 trial, where guadecitabine (30–60 mg/m2 SC, days 1–5) combined with ipilimumab resulted in a five-year overall survival rate of 28.9% [301].
3.3.4 Future Directions of Trial Design
Appropriate patient selection using validated predictive biomarkers (methylation signatures, immune phenotypes, genetic markers), optimal biological dose-finding replacing maximum tolerated dose, pharmacodynamic monitoring ensuring target engagement, and rational combinations addressing complementary resistance mechanisms can transform these agents from categorical failures into precision medicine tools for melanoma populations. Fig. 4 illustrates an algorithm of how epigenetic biomarkers are used for treatment selection, and how epigenetics agents are used as combination regimens.
3.3.5 Theoretical Biomarker-Driven Trial Schemas
Incorporating these corrective ideas, some theoretical biomarker-driven trial schemas may be: 1) Patients with high-methylation cluster tumors with IDH1 mutations may be treated with decitabine, followed by pembrolizumab. Serial ctDNA methylation monitoring can measure treatment effect, with non-responders crossing over to alternative therapy. 2) Patients with high-methylation at immune gene promoters with low PD-1 expression tumor may be treated with guadecitabine priming combined with ipilimumab then maintenance nivolumab. Serial 5-hmC increase can monitor treatment effect. 3) Patients with high HDAC2 expression with histone acetylation capacity assessment may be treated with panobinostat combined with pembrolizumab for checkpoint inhibitor rechallenge.
4 Artificial Intelligence (AI)/Integrative Multiomics Approach
AI and machine learning (ML) are revolutionizing melanoma management, particularly in biomarker discovery, patient stratification, and outcome prediction. This transformation is achieved through the integration of multi-omics data with sophisticated algorithmic techniques.
4.1 AI/ML for Biomarker Discovery
One notable approach is a transfer learning-based biomarker discovery model combined with an ensemble machine learning model. This innovative strategy successfully identified novel biomarkers, yielding an impressive AUC of 0.9861 and an overall accuracy of 91.05%. The study highlighted specific genes essential for diagnostic classification and identified both diagnostic and prognostic biomarkers specifically for melanoma [302].
4.2 AI/ML for Prognostic Stratification
In another significant advancement, researchers developed a machine learning-driven signature (MLDS) based on marker genes from various molecular subtypes. This signature notably enhanced predictive accuracy for melanoma patient prognosis. Findings indicated that MLDS scores were associated with decreased immune cell infiltration and lower expression of immune checkpoints. Patients categorized in the low MLDS group demonstrated greater responsiveness to chemotherapy and could potentially benefit more from immune checkpoint inhibitors. This study emphasizes the complexity of melanoma’s molecular subtypes and the critical role of the tumor microenvironment in disease progression [303].
4.3 ML with Multi-Omics Approach
Korfiati’s analysis utilized ML to integrate miRNA expression profiles with multi-omics, clinical, and imaging data, leading to a deeper understanding of biological processes and the identification of more informative biomarkers. The study demonstrated that miRNA signatures could be employed in training classification models to predict melanoma recurrence and metastasis with high accuracy rates of 91.51% and 97.39%, respectively. Notably, when clinical information was added, the predictive accuracy for melanoma recurrence in an external test group increased from 73.85% to 85.38% [304].
Another study employed weighted gene co-expression network analysis (WGCNA) combined with consensus clustering and 10 ML algorithms to develop an immunotherapy-related gene model (ITRGM). This multi-omics analysis, which included both bulk and single-cell RNA sequencing of melanoma patients, identified 66 consensus immunotherapy prognostic genes (CITPGs). The CITPG-high group not only exhibited improved prognosis but also showed enhanced immune activity. By utilizing a combination of ML algorithms, the ITRGM signature effectively stratified patients into high-risk or low-risk categories for immunotherapy response, serving as a reliable predictor for classifying melanoma patients into ‘immune-hot’ and ‘immune-cold’ tumors and thus enhancing immunotherapy outcomes [305].
A study focused on assessing machine learning algorithms for predicting melanoma recurrence using clinical and histopathologic data obtained from Electronic Health Records (EHRs). The study achieved recurrence classification performance with AUC values of 0.845 and 0.812, highlighting Breslow tumor thickness and mitotic rate as the most predictive features. These findings indicate that machine learning can effectively extract valuable predictive signals from clinicopathologic data, potentially leading to the identification of early-stage melanoma patients who may benefit from adjuvant immunotherapy [306].
Lastly, a recent study demonstrates that deep learning analysis of histopathology can serve as a spatiotemporal framework for optimizing the design and timing of epigenetic interventions. This approach enables the targeted elimination of resistant cancer clones, potentially improving treatment outcomes [307].
5 Challenges and Future Perspectives
Despite vast publications on melanoma epigenetics, current treatment guidelines rely solely on genetic mutations (BRAF/NRAS), tumor mutational burden, and PD-L1 expression. As of 2025, no epigenetic biomarkers have been included in standard clinical recommendations for melanoma. This significant gap between robust research and clinical use results from distinct, solvable barriers.
The main obstacle is the lack of prospective validation. Most studies on epigenetic biomarkers employ retrospective designs and small sample sizes, resulting in preliminary findings. Large, Phase III prospective trials, ideally multi-center and enrolling more than 500 patients, are needed to demonstrate that using epigenetic markers to guide treatment improves outcomes compared to standard care. While ongoing trials combining EZH2, HDAC, or DNMT inhibitors with immunotherapy or targeted therapy may eventually provide this evidence, results are still pending.
Technical standardization issues make reproducibility between labs difficult. Assay platforms, including methylation-specific Polymerase Chain Reaction (PCR), pyrosequencing, Illumina arrays, and bisulfite sequencing, often produce inconsistent results. Blood collection, storage, and processing methods also significantly affect circulating biomarker measurements.
Established biomarkers pose stiff competition, making it hard for epigenetic markers to prove additional value. Tumor mutational burden, microsatellite instability, and PD-L1 expression already inform immunotherapy choices, while BRAF and NRAS mutations determine eligibility for targeted therapy. For epigenetic markers to be adopted in clinical practice, they must either outperform current tests or offer unique, complementary information that enhances outcomes—an ambitious goal that requires direct comparison studies.
Heterogeneity of melanoma—both within individual tumors and between different metastases in the same patient—creates difficulties for all biomarker strategies, but especially for epigenetic markers, which can vary widely across tumor regions. A single biopsy may not reflect the full tumor profile, although circulating biomarkers could potentially capture this heterogeneity by sampling multiple tumor sites. Researchers have employed multi-omics approaches, including single-cell techniques, to connect epigenetic alterations to melanoma pathogenesis. A comprehensive single-cell RNA-seq analysis across multiple cancer types, including melanoma, revealed shared gene expression patterns associated with stress and differentiation. These findings have enhanced our understanding of melanoma heterogeneity and the roles of various cell states in disease advancement [308].
Complete lack of cost-effectiveness data for melanoma epigenetic biomarkers is another obstacle. Healthcare systems require evidence that these tests improve outcomes to justify their expense. Without such data, insurers will not reimburse for the tests, and clinicians are unlikely to order them—even if they become available.
The review serves as a foundation for bringing to maturity the translational realities of epigenetic approaches to melanoma therapy. Recent progress in epigenetics offers hope for improving patient outcomes by enabling melanoma management to focus on personalized strategies that integrate both genetic and epigenetic data. As evidenced in this review, the epigenetic pathways that may impact melanoma initiation, progression, and overall virulence are diverse and complex. Therefore, therapeutic approaches will need to understand how epigenetic factors collaborate in aggregate to produce and perpetuate disease in individual patients. Utilizing AI to integrate data offers promise in this regard. While cancer remains a disease of the genome, epigenetic approaches offer novel strategies for correcting the consequences of an aberrant and mutated melanoma genome.
Acknowledgement: Not applicable.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: Conceptualization, Christine G. Lian; Writing—original draft preparation, Sophiette G. Hong; Writing—review and editing, Christine G. Lian, George F. Murphy, Sophiette G. Hong. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.

References
1. Holliday R. The inheritance of epigenetic defects. Science. 1987;238(4824):163–70. doi:10.1126/science.3310230. [Google Scholar] [PubMed] [CrossRef]
2. Parry A, Rulands S, Reik W. Active turnover of DNA methylation during cell fate decisions. Nat Rev Genet. 2021;22(1):59–66. doi:10.1038/s41576-020-00287-8. [Google Scholar] [PubMed] [CrossRef]
3. Millán-Zambrano G, Burton A, Bannister AJ, Schneider R. Histone post-translational modifications—cause and consequence of genome function. Nat Rev Genet. 2022;23(9):563–80. doi:10.1038/s41576-022-00468-7. [Google Scholar] [PubMed] [CrossRef]
4. Delaunay S, Helm M, Frye M. RNA modifications in physiology and disease: towards clinical applications. Nat Rev Genet. 2024;25(2):104–22. doi:10.1038/s41576-023-00645-2. [Google Scholar] [PubMed] [CrossRef]
5. Eustermann S, Patel AB, Hopfner KP, He Y, Korber P. Energy-driven genome regulation by ATP-dependent chromatin remodellers. Nat Rev Mol Cell Biol. 2024;25(4):309–32. doi:10.1038/s41580-023-00683-y. [Google Scholar] [PubMed] [CrossRef]
6. Maia BM, Rocha RM, Calin GA. Clinical significance of the interaction between non-coding RNAs and the epigenetics machinery: challenges and opportunities in oncology. Epigenetics. 2014;9(1):75–80. doi:10.4161/epi.26488. [Google Scholar] [PubMed] [CrossRef]
7. Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20:608–24. doi:10.1038/s41580-019-0168-5. [Google Scholar] [PubMed] [CrossRef]
8. Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. 2017;49:e324. doi:10.1038/emm.2017.11. [Google Scholar] [PubMed] [CrossRef]
9. Dai W, Qiao X, Fang Y, Guo R, Bai P, Liu S, et al. Epigenetics-targeted drugs: current paradigms and future challenges. Signal Transduct Target Ther. 2024;9(1):332. doi:10.1038/s41392-024-02039-0. [Google Scholar] [PubMed] [CrossRef]
10. Karami Fath M, Azargoonjahromi A, Soofi A, Almasi F, Hosseinzadeh S, Khalili S, et al. Current understanding of epigenetics role in melanoma treatment and resistance. Cancer Cell Int. 2022;22(1):313. doi:10.1186/s12935-022-02738-0. [Google Scholar] [PubMed] [CrossRef]
11. Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–32. doi:10.1126/science.187.4173.226. [Google Scholar] [CrossRef]
12. Hermann A, Goyal R, Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem. 2004;279:48350–9. doi:10.1074/jbc.m403427200. [Google Scholar] [PubMed] [CrossRef]
13. Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23(16):5594–605. doi:10.1128/MCB.23.16.5594-5605.2003. [Google Scholar] [PubMed] [CrossRef]
14. Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5 mC to 5 hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466(7310):1129–33. doi:10.1038/nature09303. [Google Scholar] [PubMed] [CrossRef]
15. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–5. doi:10.1126/science.1170116. [Google Scholar] [PubMed] [CrossRef]
16. Szwagierczak A, Bultmann S, Schmidt CS, Spada F, Leonhardt H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010;38:e181. doi:10.1093/nar/gkq684. [Google Scholar] [PubMed] [CrossRef]
17. Lee AV, Nestler KA, Chiappinelli KB. Therapeutic targeting of DNA methylation alterations in cancer. Pharmacol Ther. 2024;258:108640. doi:10.1016/j.pharmthera.2024.108640. [Google Scholar] [PubMed] [CrossRef]
18. Yamaguchi K, Chen X, Rodgers B, Miura F, Bashtrykov P, Bonhomme F, et al. Non-canonical functions of UHRF1 maintain DNA methylation homeostasis in cancer cells. Nat Commun. 2024;15:2960. doi:10.21203/rs.3.rs-3154646/v1. [Google Scholar] [CrossRef]
19. Janic A, Abad E, Amelio I. Decoding p53 tumor suppression: a crosstalk between genomic stability and epigenetic control? Cell Death Differ. 2025;32(1):1–8. doi:10.1038/s41418-024-01259-9. [Google Scholar] [PubMed] [CrossRef]
20. Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–54. doi:10.1056/NEJMra023075. [Google Scholar] [PubMed] [CrossRef]
21. Pappalardo XG, Barra V. Losing DNA methylation at repetitive elements and breaking bad. Epigenetics Chromatin. 2021;14(1):25. doi:10.1186/s13072-021-00400-z. [Google Scholar] [PubMed] [CrossRef]
22. Besselink N, Keijer J, Vermeulen C, Boymans S, de Ridder J, Van Hoeck A, et al. The genome-wide mutational consequences of DNA hypomethylation. Sci Rep. 2023;13(1):6874. doi:10.1038/s41598-023-33932-3. [Google Scholar] [PubMed] [CrossRef]
23. Van Tongelen A, Loriot A, De Smet C. Oncogenic roles of DNA hypomethylation through the activation of cancer-germline genes. Cancer Lett. 2017;396:130–7. doi:10.1016/j.canlet.2017.03.029. [Google Scholar] [PubMed] [CrossRef]
24. Rothhammer T, Bosserhoff A-K. Epigenetic events in malignant melanoma. Pigment Cell Res. 2007;20:92–111. doi:10.1111/j.1600-0749.2007.00367.x. [Google Scholar] [PubMed] [CrossRef]
25. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–53. doi:10.1038/nrc1279. [Google Scholar] [PubMed] [CrossRef]
26. Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi:10.1038/301089a0. [Google Scholar] [PubMed] [CrossRef]
27. Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007;1775(1):138–62. doi:10.1016/j.bbcan.2006.08.007. [Google Scholar] [PubMed] [CrossRef]
28. Rollins RA, Haghighi F, Edwards JR, Das R, Zhang MQ, Ju J, et al. Large-scale structure of genomic methylation patterns. Genome Res. 2006;16(2):157–63. doi:10.1101/gr.4362006. [Google Scholar] [PubMed] [CrossRef]
29. Ecsedi SI, Hernandez-Vargas H, Lima SC, Herceg Z, Adany R, Balazs M. Transposable hypomethylation is associated with metastatic capacity of primary melanomas. Int J Clin Exp Pathol. 2013;6(12):2943–8. [Google Scholar] [PubMed]
30. Kim YI, Giuliano A, Hatch KD, Schneider A, Nour MA, Dallal GE, et al. Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma. Cancer. 1994;74(3):893–9. doi:10.1002/1097-0142(19940801)74:. [Google Scholar] [CrossRef]
31. Cravo M, Pinto R, Fidalgo P, Chaves P, Glória L, Nobre-Leitão C, et al. Global DNA hypomethylation occurs in the early stages of intestinal type gastric carcinoma. Gut. 1996;39(3):434–8. doi:10.1136/gut.39.3.434. [Google Scholar] [PubMed] [CrossRef]
32. Meevassana J, Varophas S, Prabsattru P, Kamolratanakul S, Ruangritchankul K, Kitkumthorn N. 5-Methylcytosine immunohistochemistry for predicting cutaneous melanoma prognosis. Sci Rep. 2024;14(1):7554. doi:10.1038/s41598-024-58011-z. [Google Scholar] [PubMed] [CrossRef]
33. Micevic G, Theodosakis N, Taube JM, Bosenberg MW, Rodić N. Attenuation of genome-wide 5-methylcytosine level is an epigenetic feature of cutaneous malignant melanomas. Melanoma Res. 2017;27(2):85–96. doi:10.1097/CMR.0000000000000315. [Google Scholar] [PubMed] [CrossRef]
34. Traynor S, Jakobsen MK, Green TM, Komic H, Palarasah Y, Pedersen CB, et al. Single-cell sequencing unveils extensive intratumoral heterogeneity of cancer/testis antigen expression in melanoma and lung cancer. J Immunother Cancer. 2024;12(6):e008759. doi:10.1136/jitc-2023-008759. [Google Scholar] [PubMed] [CrossRef]
35. Schinke C, Mo Y, Yu Y, Amiri K, Sosman J, Greally J, et al. Aberrant DNA methylation in malignant melanoma. Melanoma Res. 2010;20(4):253–65. doi:10.1097/CMR.0b013e328338a35a. [Google Scholar] [PubMed] [CrossRef]
36. Zendman AJ, de Wit NJ, Van Kraats AA, Weidle UH, Ruiter DJ, Van Muijen GN. Expression profile of genes coding for melanoma differentiation antigens and cancer/testis antigens in metastatic lesions of human cutaneous melanoma. Melanoma Res. 2001;11(5):451–9. doi:10.1097/00008390-200110000-00003. [Google Scholar] [PubMed] [CrossRef]
37. Li S, Shi X, Li J, Zhou X. Pathogenicity of the MAGE family. Oncol Lett. 2021;22(6):844. doi:10.3892/ol.2021.13105. [Google Scholar] [PubMed] [CrossRef]
38. Forchhammer S, Pop OT, Hahn M, Aebischer V, Seitz CM, Schroeder C, et al. Expression of the tumor antigens NY-ESO-1, tyrosinase, MAGE-A3, and TPTE in pediatric and adult melanoma: a retrospective case control study. Virchows Arch. 2024;485(2):335–46. doi:10.1007/s00428-024-03846-0. [Google Scholar] [PubMed] [CrossRef]
39. Aung PP, Liu YC, Ballester LY, Robbins PF, Rosenberg SA, Lee CC. Expression of New York esophageal squamous cell carcinoma-1 in primary and metastatic melanoma. Hum Pathol. 2014;45(2):259–67. doi:10.1016/j.humpath.2013.05.029. [Google Scholar] [PubMed] [CrossRef]
40. Olvedy M, Tisserand JC, Luciani F, Boeckx B, Wouters J, Lopez S, et al. Comparative oncogenomics identifies tyrosine kinase FES as a tumor suppressor in melanoma. J Clin Invest. 2017;127(6):2310–25. doi:10.1172/JCI91291. [Google Scholar] [PubMed] [CrossRef]
41. Venza M, Visalli M, Catalano T, Beninati C, Teti D, Venza I. DSS1 promoter hypomethylation and overexpression predict poor prognosis in melanoma and squamous cell carcinoma patients. Hum Pathol. 2017;60:137–46. doi:10.1016/j.humpath.2016.10.018. [Google Scholar] [PubMed] [CrossRef]
42. Jia J, Gu J, Gao L, Liu K, Dai J, Zhang F, et al. CNDP1 overexpression by promoter hypomethylation predicts poor prognosis and immunotherapy response in mucosal melanoma. Cancer Sci. 2025;116(6):1671–8. doi:10.1111/cas.70062. [Google Scholar] [PubMed] [CrossRef]
43. Vizoso M, Ferreira HJ, Lopez-Serra P, Carmona FJ, Martínez-Cardús A, Girotti MR, et al. Epigenetic activation of a cryptic TBC1D16 transcript enhances melanoma progression by targeting EGFR. Nat Med. 2015;21(7):741–50. doi:10.1038/nm.3863. [Google Scholar] [PubMed] [CrossRef]
44. Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K, et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150(6):1135–46. doi:10.1016/j.cell.2012.07.033. [Google Scholar] [PubMed] [CrossRef]
45. Hao J, Huang Z, Zhang S, Song K, Wang J, Gao C, et al. Deciphering the multifaceted roles and clinical implications of 2-hydroxyglutarate in cancer. Pharmacol Res. 2024;209:107437. doi:10.1016/j.phrs.2024.107437. [Google Scholar] [PubMed] [CrossRef]
46. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-Ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi:10.1016/j.ccr.2010.12.014. [Google Scholar] [PubMed] [CrossRef]
47. Lee JJ, Cook M, Mihm MC, Xu S, Zhan Q, Wang TJ, et al. Loss of the epigenetic mark, 5-Hydroxymethylcytosine, correlates with small cell/nevoid subpopulations and assists in microstaging of human melanoma. Oncotarget. 2015;6(35):37995–8004. doi:10.18632/oncotarget.6062. [Google Scholar] [PubMed] [CrossRef]
48. Bonvin E, Radaelli E, Bizet M, Luciani F, Calonne E, Putmans P, et al. TET2-dependent hydroxymethylome plasticity reduces melanoma initiation and progression. Cancer Res. 2019;79(3):482–94. doi:10.1158/0008-5472.CAN-18-1214. [Google Scholar] [PubMed] [CrossRef]
49. Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6(5):963–8. doi:10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [Google Scholar] [PubMed] [CrossRef]
50. Vanharanta S, Massagué J. Field cancerization: something new under the sun. Cell. 2012;149(6):1179–81. doi:10.1016/j.cell.2012.05.013. [Google Scholar] [PubMed] [CrossRef]
51. Larson AR, Dresser KA, Zhan Q, Lezcano C, Woda BA, Yosufi B, et al. Loss of 5-hydroxymethylcytosine correlates with increasing morphologic dysplasia in melanocytic tumors. Mod Pathol. 2014;27(7):936–44. doi:10.1038/modpathol.2013.224. [Google Scholar] [PubMed] [CrossRef]
52. Gambichler T, Sand M, Skrygan M. Loss of 5-hydroxymethylcytosine and ten-eleven translocation 2 protein expression in malignant melanoma. Melanoma Res. 2013;23(3):218–20. doi:10.1097/CMR.0b013e32835f9bd4. [Google Scholar] [PubMed] [CrossRef]
53. Siref AB, Huynh CAT, Balzer BL, Frishberg DP, Essner R, Shon W. Diagnostic utility of dual 5-hydroxymethylcytosine/Melan-A immunohistochemistry in differentiating nodal nevus from metastatic melanoma: an effective first-line test for the workup of sentinel lymph node specimen. J Cutan Pathol. 2019;46(4):261–6. doi:10.1111/cup.13412. [Google Scholar] [PubMed] [CrossRef]
54. Fang R, Vallius T, Zhang A, Van Cura D, Alicandri F, Fischer G, et al. PRAME expression in melanoma is negatively regulated by TET2-mediated DNA hydroxymethylation. Lab Invest. 2025;105(5):104123. doi:10.1016/j.labinv.2025.104123. [Google Scholar] [PubMed] [CrossRef]
55. Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018 Jun;19(6):371–84. doi:10.1038/s41576-018-0004-3. PMID: 29643443. [Google Scholar] [PubMed] [CrossRef]
56. Lee JJ, Murphy GF, Lian CG. Melanoma epigenetics: novel mechanisms, markers, and medicines. Lab Invest. 2014;94:822–38. doi:10.1038/labinvest.2014.87. [Google Scholar] [PubMed] [CrossRef]
57. Aleotti V, Catoni C, Poggiana C, Rosato A, Facchinetti A, Scaini MC. Methylation markers in cutaneous melanoma: unravelling the potential utility of their tracking by liquid biopsy. Cancers. 2021;13(24):6217. doi:10.3390/cancers13246217. [Google Scholar] [PubMed] [CrossRef]
58. Fu S, Wu H, Zhang H, Lian CG, Lu Q. DNA methylation/hydroxymethylation in melanoma. Oncotarget. 2017;8(44):78163–73. doi:10.18632/oncotarget.18293. [Google Scholar] [PubMed] [CrossRef]
59. Davalos V, Lovell CD, Von Itter R, Dolgalev I, Agrawal P, Baptiste G, et al. An epigenetic switch controls an alternative NR2F2 isoform that unleashes a metastatic program in melanoma. Nat Commun. 2023;14(1):1867. doi:10.1038/s41467-023-36967-2. [Google Scholar] [PubMed] [CrossRef]
60. Akinleye A, Avvaru P, Furqan M, Song Y, Liu D. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J Hematol Oncol. 2013;6(1):88. doi:10.1186/1756-8722-6-88. [Google Scholar] [PubMed] [CrossRef]
61. Gào X, Schöttker B. Reduction-oxidation pathways involved in cancer development: a systematic review of literature reviews. Oncotarget. 2017;8(31):51888–906. doi:10.18632/oncotarget.17128. [Google Scholar] [PubMed] [CrossRef]
62. Mirmohammadsadegh A, Marini A, Nambiar S, Hassan M, Tannapfel A, Ruzicka T, et al. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res. 2006;66(13):6546–52. doi:10.1158/0008-5472.can-06-0384. [Google Scholar] [PubMed] [CrossRef]
63. Lahtz C, Stranzenbach R, Fiedler E, Helmbold P, Dammann RH. Methylation of PTEN as a prognostic factor in malignant melanoma of the skin. J Invest Dermatol. 2010;130(2):620–2. doi:10.1038/jid.2009.226. [Google Scholar] [PubMed] [CrossRef]
64. Giles KM, Rosenbaum BE, Berger M, Izsak A, Li Y, Illa Bochaca I, et al. Revisiting the clinical and biologic relevance of partial PTEN loss in melanoma. J Invest Dermatol. 2019;139(2):430–8. doi:10.1016/j.jid.2018.07.031. [Google Scholar] [PubMed] [CrossRef]
65. Zeng H, Jorapur A, Shain AH, Lang UE, Torres R, Zhang Y, et al. Bi-allelic loss of CDKN2A initiates melanoma invasion via BRN2 activation. Cancer Cell. 2018;34(1):56–68.e9. doi:10.1016/j.ccell.2018.05.014. [Google Scholar] [PubMed] [CrossRef]
66. Van der Velden PA, Metzelaar-Blok JA, Bergman W, Monique H, Hurks H, Frants RR, et al. Promoter hypermethylation: a common cause of reduced p16INK4a expression in uveal melanoma. Can Res. 2001;61(13):5303–6. [Google Scholar]
67. Ming Z, Lim SY, Rizos H. Genetic alterations in the INK4a/ARF locus: effects on melanoma development and progression. Biomolecules. 2020;10(10):1447. doi:10.3390/biom10101447. [Google Scholar] [PubMed] [CrossRef]
68. Venza M, Visalli M, Biondo C, Lentini M, Catalano T, Teti D, et al. Epigenetic regulation of p14ARF and p16INK4A expression in cutaneous and uveal melanoma. Biochim Biophys Acta. 2015;1849(3):247–56. doi:10.1016/j.bbagrm.2014.12.004. [Google Scholar] [PubMed] [CrossRef]
69. Micevic G, Theodosakis N, Bosenberg M. Aberrant DNA methylation in melanoma: biomarker and therapeutic opportunities. Clin Epigenetics. 2017;9:34. doi:10.1186/s13148-017-0332-8. [Google Scholar] [PubMed] [CrossRef]
70. Straume O, Smeds J, Kumar R, Hemminki K, Akslen LA. Significant impact of promoter hypermethylation and the 540 C>T polymorphism of CDKN2A in cutaneous melanoma of the vertical growth phase. Am J Pathol. 2002;161(1):229–37. doi:10.1016/S0002-9440(10)64174-0. [Google Scholar] [PubMed] [CrossRef]
71. Pfeifer GP, Yoon JH, Liu L, Tommasi S, Wilczynski SP, Dammann R. Methylation of the RASSF1A gene in human cancers. Biol Chem. 2002;383(6):907–14. doi:10.1515/BC.2002.097. [Google Scholar] [PubMed] [CrossRef]
72. Spugnardi M, Tommasi S, Dammann R, Pfeifer GP, Hoon DS. Epigenetic inactivation of RAS association domain family protein 1 (RASSF1A) in malignant cutaneous melanoma. Cancer Res. 2003;63(7):1639–43. [Google Scholar] [PubMed]
73. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339(6122):959–61. doi:10.1126/science.1230062. [Google Scholar] [PubMed] [CrossRef]
74. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339(6122):957–9. doi:10.1126/science.1229259. [Google Scholar] [PubMed] [CrossRef]
75. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110(15):6021–6. doi:10.1073/pnas.1303607110. [Google Scholar] [PubMed] [CrossRef]
76. Vinagre J, Almeida A, Pópulo H, Batista R, Lyra J, Pinto V, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185. doi:10.1038/ncomms3185. [Google Scholar] [PubMed] [CrossRef]
77. Lee DD, Leão R, Komosa M, Gallo M, Zhang CH, Lipman T, et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J Clin Invest. 2019;129(1):223–9. doi:10.1172/JCI121303. [Google Scholar] [PubMed] [CrossRef]
78. Salgado C, Roelse C, Nell R, Gruis N, Van Doorn R, Van der Velden P. Interplay between TERT promoter mutations and methylation culminates in chromatin accessibility and TERT expression. PLoS One. 2020;15(4):e0231418. doi:10.1371/journal.pone.0231418. [Google Scholar] [PubMed] [CrossRef]
79. Seynnaeve B, Lee S, Borah S, Park Y, Pappo A, Kirkwood JM, et al. Genetic and epigenetic alterations of TERT are associated with inferior outcome in adolescent and young adult patients with melanoma. Sci Rep. 2017;7:45704. doi:10.1038/srep45704. [Google Scholar] [PubMed] [CrossRef]
80. Tuominen R, Jewell R, Van den Oord JJ, Wolter P, Stierner U, Lindholm C, et al. MGMT promoter methylation is associated with temozolomide response and prolonged progression-free survival in disseminated cutaneous melanoma. Int J Cancer. 2015;136(12):2844–53. doi:10.1002/ijc.29332. [Google Scholar] [PubMed] [CrossRef]
81. Yun HS, Kramp TR, Palanichamy K, Tofilon PJ, Camphausen K. MGMT inhibition regulates radioresponse in GBM, GSC, and melanoma. Sci Rep. 2024;14(1):12363. doi:10.1038/s41598-024-61240-x. [Google Scholar] [PubMed] [CrossRef]
82. Bernstein E, Hake SB. The nucleosome: a little variation goes a long way. Biochem Cell Biol. 2006;84(4):505–17. doi:10.1139/o06-085. [Google Scholar] [PubMed] [CrossRef]
83. Vardabasso C, Hasson D, Ratnakumar K, Chung CY, Duarte LF, Bernstein E. Histone variants: emerging players in cancer biology. Cell Mol Life Sci. 2014;71(3):379–404. doi:10.1007/s00018-013-1343-z. [Google Scholar] [PubMed] [CrossRef]
84. Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20(3):259–66. doi:10.1038/nsmb.2470. [Google Scholar] [PubMed] [CrossRef]
85. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128(4):635–8. doi:10.1016/j.cell.2007.02.006. [Google Scholar] [PubMed] [CrossRef]
86. Cutter AR, Hayes JJ. A brief review of nucleosome structure. FEBS Lett. 2015;589(20PtA):2914–22. doi:10.1016/j.febslet.2015.05.016. [Google Scholar] [PubMed] [CrossRef]
87. Dueva R, Akopyan K, Pederiva C, Trevisan D, Dhanjal S, Lindqvist A, et al. Neutralization of the positive charges on histone tails by RNA promotes an open chromatin structure. Cell Chem Biol. 2019;26(10):1436–49.e5. doi:10.1016/j.chembiol.2019.08.002. [Google Scholar] [PubMed] [CrossRef]
88. Nitsch S, Zorro Shahidian L, Schneider R. Histone acylations and chromatin dynamics: concepts, challenges, and links to metabolism. EMBO Rep. 2021;22(7):e52774. doi:10.15252/embr.202152774. [Google Scholar] [PubMed] [CrossRef]
89. Xu L, Xuan H, Shi X. Dysregulation of the p300/CBP histone acetyltransferases in human cancer. Epigenomics. 2025;17(3):193–208. doi:10.1080/17501911.2024.2447807. [Google Scholar] [PubMed] [CrossRef]
90. Wang R, He Y, Robinson V, Yang Z, Hessler P, Lasko LM, et al. Targeting lineage-specific MITF pathway in human melanoma cell lines by A-485, the selective small-molecule inhibitor of p300/CBP. Mol Cancer Ther. 2018;17(12):2543–50. doi:10.1158/1535-7163.MCT-18-0511. [Google Scholar] [PubMed] [CrossRef]
91. Wu X, Zhang X, Tang S, Wang Y. The important role of the histone acetyltransferases p300/CBP in cancer and the promising anticancer effects of p300/CBP inhibitors. Cell Biol Toxicol. 2025;17, 41(1):32. doi:10.1007/s10565-024-09984-0. [Google Scholar] [PubMed] [CrossRef]
92. Waddell A, Grbic N, Leibowitz K, Wyant WA, Choudhury S, Park K, et al. p300 KAT regulates SOX10 stability and function in human melanoma. Cancer Res Commun. 2024;4(8):1894–907. doi:10.1158/2767-9764.CRC-24-0124. [Google Scholar] [PubMed] [CrossRef]
93. Strub T, Ballotti R, Bertolotto C. The “ART” of epigenetics in melanoma: from histone “alterations, to resistance and therapies”. Theranostics. 2020;10(4):1777–97. doi:10.7150/thno.36218. [Google Scholar] [PubMed] [CrossRef]
94. Segura MF, Fontanals-Cirera B, Gaziel-Sovran A, Guijarro MV, Hanniford D, Zhang G, et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013;73(20):6264–76. doi:10.1158/0008-5472.CAN-13-0122-T. [Google Scholar] [PubMed] [CrossRef]
95. Bergman JA, Woan K, Perez-Villarroel P, Villagra A, Sotomayor EM, Kozikowski AP. Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J Med Chem. 2012;55:9891–9. doi:10.1021/jm301098e. [Google Scholar] [PubMed] [CrossRef]
96. Woan KV, Lienlaf M, Perez-Villaroel P, Lee C, Cheng F, Knox T, et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: enhanced antitumor immunity and impaired cell proliferation. Mol Oncol. 2015;9(7):1447–57. doi:10.1016/j.molonc.2015.04.002. [Google Scholar] [PubMed] [CrossRef]
97. Vance KW, Carreira S, Brosch G, Goding CR. Tbx2 is overexpressed and plays an important role in maintaining proliferation and suppression of senescence in melanomas. Cancer Res. 2005;65(6):2260–8. doi:10.1158/0008-5472.CAN-04-3045. [Google Scholar] [PubMed] [CrossRef]
98. Wilmott JS, Colebatch AJ, Kakavand H, Shang P, Carlino MS, Thompson JF, et al. Expression of the class 1 histone deacetylases HDAC8 and 3 are associated with improved survival of patients with metastatic melanoma. Mod Pathol. 2015;28(7):884–94. doi:10.1038/modpathol.2015.34. [Google Scholar] [PubMed] [CrossRef]
99. Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol. 2021;12:636568. doi:10.3389/fimmu.2021.636568. [Google Scholar] [PubMed] [CrossRef]
100. Serrano A, Castro-Vega I, Redondo M. Role of gene methylation in antitumor immune response: implication for tumor progression. Cancers. 2011;3(2):1672–90. doi:10.3390/cancers3021672. [Google Scholar] [PubMed] [CrossRef]
101. Xu P, Xiong W, Lin Y, Fan L, Pan H, Li Y. Histone deacetylase 2 knockout suppresses immune escape of triple-negative breast cancer cells via downregulating PD-L1 expression. Cell Death Dis. 2021;12(8):779. doi:10.1038/s41419-021-04047-2. [Google Scholar] [PubMed] [CrossRef]
102. Peinado H, Ballestar E, Esteller M, Cano A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol. 2004;24(1):306–19. doi:10.1128/MCB.24.1.306-319.2004. [Google Scholar] [PubMed] [CrossRef]
103. Aghdassi A, Sendler M, Guenther A, Mayerle J, Behn CO, Heidecke CD, et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. 2012;61(3):439–48. doi:10.1136/gutjnl-2011-300060. [Google Scholar] [PubMed] [CrossRef]
104. Song C, Zhu S, Wu C, Kang J. Histone deacetylase (HDAC) 10 suppresses cervical cancer metastasis through inhibition of matrix metalloproteinase (MMP) 2 and 9 expression. J Biol Chem. 2013;288(39):28021–33. doi:10.1074/jbc.M113.498758. [Google Scholar] [PubMed] [CrossRef]
105. Emmons MF, Bennett RL, Riva A, Gupta K, Carvalho LADC, Zhang C, et al. HDAC8-mediated inhibition of EP300 drives a transcriptional state that increases melanoma brain metastasis. Nat Commun. 2023;14(1):7759. doi:10.1038/s41467-023-43519-1. [Google Scholar] [PubMed] [CrossRef]
106. Orouji E, Utikal J. Tackling malignant melanoma epigenetically: histone lysine methylation. Clin Epigenetics. 2018;10(1):145. doi:10.1186/s13148-018-0583-z. [Google Scholar] [PubMed] [CrossRef]
107. Duan X, Xing Z, Qiao L, Qin S, Zhao X, Gong Y, et al. The role of histone post-translational modifications in cancer and cancer immunity: functions, mechanisms and therapeutic implications. Front Immunol. 2024;15:1495221. doi:10.3389/fimmu.2024.1495221. [Google Scholar] [PubMed] [CrossRef]
108. Zingg D, Debbache J, Schaefer SM, Tuncer E, Frommel SC, Cheng P, et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. 2015;6:6051. doi:10.1038/ncomms7051. [Google Scholar] [PubMed] [CrossRef]
109. Mahmoud F, Shields B, Makhoul I, Hutchins LF, Shalin SC, Tackett AJ. Role of EZH2 histone methyltrasferase in melanoma progression and metastasis. Cancer Biol Ther. 2016;17(6):579–91. doi:10.1080/15384047.2016.1167291. [Google Scholar] [PubMed] [CrossRef]
110. Kang N, Eccleston M, Clermont PL, Latarani M, Male DK, Wang Y, et al. EZH2 inhibition: a promising strategy to prevent cancer immune editing. Epigenomics. 2020;12(16):1457–76. doi:10.2217/epi-2020-0186. [Google Scholar] [PubMed] [CrossRef]
111. Zhu CY, Zhai TT, Su M, Pan HC, Tang Q, Huang BH, et al. EZH2 elicits CD8+ T-cell desert in esophageal squamous cell carcinoma via suppressing CXCL9 and dendritic cells. Commun Biol. 2024;7(1):1645. doi:10.1038/s42003-024-07341-9. [Google Scholar] [PubMed] [CrossRef]
112. Li Z, Wang D, Lu J, Huang B, Wang Y, Dong M, et al. Methylation of EZH2 by PRMT1 regulates its stability and promotes breast cancer metastasis. Cell Death Differ. 2020;27(12):3226–42. doi:10.1038/s41418-020-00615-9. [Google Scholar] [PubMed] [CrossRef]
113. Kostaki M, Manona AD, Stavraka I, Korkolopoulou P, Levidou G, Trigka EA, et al. High-frequency p16INK4A promoter methylation is associated with histone methyltransferase SETDB1 expression in sporadic cutaneous melanoma. Exp Dermatol. 2014;23(5):332–8. doi:10.1111/exd.12398. [Google Scholar] [PubMed] [CrossRef]
114. Orouji E, Federico A, Larribère L, Novak D, Lipka DB, Assenov Y, et al. Histone methyltransferase SETDB1 contributes to melanoma tumorigenesis and serves as a new potential therapeutic target. Int J Cancer. 2019;145(12):3462–77. doi:10.1002/ijc.32432. [Google Scholar] [PubMed] [CrossRef]
115. Cuellar TL, Herzner AM, Zhang X, Goyal Y, Watanabe C, Friedman BA, et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J Cell Biol. 2017;216(11):3535–49. doi:10.1083/jcb.201612160. [Google Scholar] [PubMed] [CrossRef]
116. Zhu B, Chen S, Wang H, Yin C, Han C, Peng C, et al. The protective role of DOT1L in UV-induced melanomagenesis. Nat Commun. 2018;9(1):259. doi:10.1038/s41467-017-02687-7. [Google Scholar] [PubMed] [CrossRef]
117. Guo C, Chen LH, Huang Y, Chang CC, Wang P, Pirozzi CJ, et al. KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget. 2013;4(11):2144–53. doi:10.18632/oncotarget.1555. [Google Scholar] [PubMed] [CrossRef]
118. Bossi D, Cicalese A, Dellino GI, Luzi L, Riva L, D’Alesio C, et al. In vivo genetic screens of patient-derived tumors revealed unexpected frailty of the transformed phenotype. Cancer Discov. 2016;6(6):650–63. doi:10.1158/2159-8290.CD-15-1200. [Google Scholar] [PubMed] [CrossRef]
119. Li L, Zhang Z, Ma T, Huo R. PRMT1 regulates tumor growth and metastasis of human melanoma via targeting ALCAM. Mol Med Rep. 2016;14(1):521–8. doi:10.3892/mmr.2016.5273. [Google Scholar] [PubMed] [CrossRef]
120. Kuźbicki L, Lange D, Strączyńska-Niemiec A, Chwirot BW. JARID1B expression in human melanoma and benign melanocytic skin lesions. Melanoma Res. 2013;23(1):8–12. doi:10.1097/CMR.0b013e32835d5d6f. [Google Scholar] [PubMed] [CrossRef]
121. Chauvistré H, Shannan B, Daignault-Mill SM, Ju RJ, Picard D, Egetemaier S, et al. Persister state-directed transitioning and vulnerability in melanoma. Nat Commun. 2022;13(1):3055. doi:10.1038/s41467-022-30641-9. [Google Scholar] [PubMed] [CrossRef]
122. Park WY, Hong BJ, Lee J, Choi C, Kim MY. H3K27 demethylase JMJD3 employs the NF-κB and BMP signaling pathways to modulate the tumor microenvironment and promote melanoma progression and metastasis. Cancer Res. 2016;76(1):161–70. doi:10.1158/0008-5472.CAN-15-0536. [Google Scholar] [PubMed] [CrossRef]
123. Yu Y, Schleich K, Yue B, Ji S, Lohneis P, Kemper K, et al. Targeting the senescence-overriding cooperative activity of structurally unrelated H3K9 demethylases in melanoma. Cancer Cell. 2018;33(2):322–336.e8. doi:10.1016/j.ccell.2018.01.002. Erratum in: Cancer Cell. 2018 Apr 9;33(4):785. doi: 10.1016/j.ccell.2018.03.009. [Google Scholar] [PubMed] [CrossRef]
124. Punnia-Moorthy G, Hersey P, Emran AA, Tiffen J. Lysine demethylases: promising drug targets in melanoma and other cancers. Front Genet. 2021;12:680633. doi:10.3389/fgene.2021.680633. [Google Scholar] [PubMed] [CrossRef]
125. Li Y, Jia R, Ge S. Role of epigenetics in uveal melanoma. Int J Biol Sci. 2017;13(4):426–33. doi:10.7150/ijbs.18331. [Google Scholar] [PubMed] [CrossRef]
126. Zhang K, Dent SY. Histone modifying enzymes and cancer: going beyond histones. J Cell Biochem. 2005;96(6):1137–48. doi:10.1002/jcb.20615. [Google Scholar] [PubMed] [CrossRef]
127. Soysouvanh F, Giuliano S, Habel N, El-Hachem N, Pisibon C, Bertolotto C, et al. An update on the role of ubiquitination in melanoma development and therapies. J Clin Med. 2021;10(5):1133. doi:10.3390/jcm10051133. [Google Scholar] [PubMed] [CrossRef]
128. Ma J, Guo W, Li C. Ubiquitination in melanoma pathogenesis and treatment. Cancer Med. 2017;6(6):1362–77. doi:10.1002/cam4.1069. [Google Scholar] [PubMed] [CrossRef]
129. Montagnani V, Maresca L, Apollo A, Pepe S, Carr RM, Fernandez-Zapico ME, et al. E3 ubiquitin ligase PARK2, an inhibitor of melanoma cell growth, is repressed by the oncogenic ERK1/2-ELK1 transcriptional axis. J Biol Chem. 2020;295(47):16058–71. doi:10.1074/jbc.RA120.014615. [Google Scholar] [PubMed] [CrossRef]
130. Yang Q, Zhao J, Chen D, Wang Y. E3 ubiquitin ligases: styles, structures and functions. Mol Biomed. 2021;2(1):23. doi:10.1186/s43556-021-00043-2. [Google Scholar] [PubMed] [CrossRef]
131. Deng L, Meng T, Chen L, Wei W, Wang P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Target Ther. 2020;5(1):11. doi:10.1038/s41392-020-0107-0. [Google Scholar] [PubMed] [CrossRef]
132. Yao W, Hu X, Wang X. Crossing epigenetic frontiers: the intersection of novel histone modifications and diseases. Signal Transduct Target Ther. 2024;9(1):232. doi:10.1038/s41392-024-01918-w. [Google Scholar] [PubMed] [CrossRef]
133. Xu Y, Zhang H, Nie D. Histone modifications and metabolic reprogramming in tumor-associated macrophages: a potential target of tumor immunotherapy. Front Immunol. 2025;16:1521550. doi:10.3389/fimmu.2025.1521550. [Google Scholar] [PubMed] [CrossRef]
134. Suskiewicz MJ, Prokhorova E, Rack JGM, Ahel I. ADP-ribosylation from molecular mechanisms to therapeutic implications. Cell. 2023;186(21):4475–95. doi:10.1016/j.cell.2023.08.030. [Google Scholar] [PubMed] [CrossRef]
135. Yu J, Chai P, Xie M, Ge S, Ruan J, Fan X, et al. Histone lactylation drives oncogenesis by facilitating m6A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 2021;22(1):85. doi:10.1186/s13059-021-02308-z. [Google Scholar] [PubMed] [CrossRef]
136. Giblin W, Bringman-Rodenbarger L, Guo AH, Kumar S, Monovich AC, Mostafa AM, et al. The deacylase SIRT5 supports melanoma viability by influencing chromatin dynamics. J Clin Invest. 2021;131(12):e138926. doi:10.1172/JCI138926. [Google Scholar] [PubMed] [CrossRef]
137. Meng F, Yuan Y, Ren H, Yue H, Xu B, Qian J. SUMOylation regulates Rb hyperphosphorylation and inactivation in uveal melanoma. Cancer Sci. 2022;113(2):622–33. doi:10.1111/cas.15223. [Google Scholar] [PubMed] [CrossRef]
138. Leucci E, Vendramin R, Spinazzi M, Laurette P, Fiers M, Wouters J, et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature. 2016;531(7595):518–22. doi:10.1038/nature17161. [Google Scholar] [PubMed] [CrossRef]
139. Bossaghzadeh F, Hajjari M, Sheikhi A, Salahshourifar I, Irani S. HOTAIR Induces the downregulation of miR-200 family members in gastric cancer cell lines. Iran Biomed J. 2022;26(1):77–84. doi:10.52547/ibj.26.1.77. [Google Scholar] [PubMed] [CrossRef]
140. Zhang D, Pei S, Feng Z, Xia G. Functions and mechanisms of lncRNAs in immune escape and their application in immunotherapy for colorectal cancer. J Transl Med. 2025;23(1):689. doi:10.1186/s12967-025-06732-8. [Google Scholar] [PubMed] [CrossRef]
141. Giunta EF, Arrichiello G, Curvietto M, Pappalardo A, Bosso D, Rosanova M, et al. Epigenetic regulation in melanoma: facts and hopes. Cells. 2021;10(8):2048. doi:10.3390/cells10082048. [Google Scholar] [PubMed] [CrossRef]
142. Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294(5543):853–8. doi:10.1126/science.1064921. [Google Scholar] [PubMed] [CrossRef]
143. Costa FF. Non-coding RNAs: lost in translation? Gene. 2007;386(1–2):1–10. doi:10.1016/j.gene.2006.09.028. [Google Scholar] [PubMed] [CrossRef]
144. Liu B, Li J, Cairns MJ. Identifying miRNAs, targets and functions. Brief Bioinform. 2014;15(1):1–19. doi:10.1093/bib/bbs075. [Google Scholar] [PubMed] [CrossRef]
145. Lim SY, Boyd SC, Diefenbach RJ, Rizos H. Circulating MicroRNAs: functional biomarkers for melanoma prognosis and treatment. Mol Cancer. 2025;24(1):99. doi:10.1186/s12943-025-02298-7. [Google Scholar] [PubMed] [CrossRef]
146. Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9(3):219–30. doi:10.1038/nrm2347. [Google Scholar] [PubMed] [CrossRef]
147. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97. doi:10.1016/s0092-8674(04)00045-5. [Google Scholar] [PubMed] [CrossRef]
148. Ye Q, Li Z, Li Y, Li Y, Zhang Y, Gui R, et al. Exosome-derived microRNA: implications in melanoma progression, diagnosis and treatment. Cancers. 2022;15(1):80. doi:10.3390/cancers15010080. [Google Scholar] [PubMed] [CrossRef]
149. Li Z, Gao Y, Cao Y, He F, Jiang R, Liu H, et al. Extracellular RNA in melanoma: advances, challenges, and opportunities. Front Cell Dev Biol. 2023;11:1141543. doi:10.3389/fcell.2023.1141543. [Google Scholar] [PubMed] [CrossRef]
150. Vignard V, Labbé M, Marec N, André-Grégoire G, Jouand N, Fonteneau JF, et al. MicroRNAs in tumor exosomes drive immune escape in melanoma. Cancer Immunol Res. 2020;8(2):255–67. doi:10.1158/2326-6066.CIR-19-0522. [Google Scholar] [PubMed] [CrossRef]
151. Luo C, Weber CE, Osen W, Bosserhoff AK, Eichmüller SB. The role of microRNAs in melanoma. Eur J Cell Biol. 2014 Jan-Feb;93(1–2):11–22. doi:10.1016/j.ejcb.2014.02.001. [Google Scholar] [PubMed] [CrossRef]
152. Poniewierska-Baran A, Słuczanowska-Głąbowska S, Małkowska P, Sierawska O, Zadroga Ł, Pawlik A, et al. Role of miRNA in melanoma development and progression. Int J Mol Sci. 2022;24(1):201. doi:10.3390/ijms24010201. [Google Scholar] [PubMed] [CrossRef]
153. Rhim J, Baek W, Seo Y, Kim JH. From molecular mechanisms to therapeutics: understanding MicroRNA-21 in cancer. Cells. 2022;11(18):2791. doi:10.3390/cells11182791. [Google Scholar] [PubMed] [CrossRef]
154. Melnik BC, John SM, Carrera-Bastos P, Schmitz G. MicroRNA-21-Enriched exosomes as epigenetic regulators in melanomagenesis and melanoma progression: the impact of western lifestyle factors. Cancers. 2020;12(8):2111. doi:10.3390/cancers12082111. [Google Scholar] [PubMed] [CrossRef]
155. Di Martino MT, Arbitrio M, Caracciolo D, Cordua A, Cuomo O, Grillone K, et al. miR-221/222 as biomarkers and targets for therapeutic intervention on cancer and other diseases: a systematic review. Mol Ther Nucleic Acids. 2022;27:1191–224. doi:10.1016/j.omtn.2022.02.005. [Google Scholar] [PubMed] [CrossRef]
156. Andreucci E, Ruzzolini J, Bianchini F, Versienti G, Biagioni A, Lulli M, et al. miR-214-enriched extracellular vesicles released by acid-adapted melanoma cells promote inflammatory macrophage-dependent tumor trans-endothelial migration. Cancers. 2022;14(20):5090. doi:10.3390/cancers14205090. [Google Scholar] [PubMed] [CrossRef]
157. Masoumeh H, Tunay D, Demet ÖA, Samuray T, Hülya Y. Exploring of miR-155-5p, miR-181b-5p, and miR-454-3p Expressions in circulating cell-free RNA: insights from peripheral blood of uveal malignant melanoma patients. Biochem Genet. 2025;63(4):3187–205. doi:10.1007/s10528-024-10849-8. [Google Scholar] [PubMed] [CrossRef]
158. Guo Y, Shi W, Fang R. miR-18a-5p promotes melanoma cell proliferation and inhibits apoptosis and autophagy by targeting EPHA7 signaling. Mol Med Rep. 2021;23(1):79. doi:10.3892/mmr.2020.11717. [Google Scholar] [PubMed] [CrossRef]
159. Liu S, Tetzlaff MT, Wang T, Yang R, Xie L, Zhang G, et al. miR-200c/Bmi1 axis and epithelial-mesenchymal transition contribute to acquired resistance to BRAF inhibitor treatment. Pigment Cell Melanoma Res. 2015;28(4):431–41. doi:10.1111/pcmr.12379. [Google Scholar] [PubMed] [CrossRef]
160. Sánchez-Sendra B, Serna E, Navarro L, González-Muñoz JF, Portero J, Ramos A, et al. Transcriptomic identification of miR-205 target genes potentially involved in metastasis and survival of cutaneous malignant melanoma. Sci Rep. 2020;10(1):4771. doi:10.1038/s41598-020-61637-4. [Google Scholar] [PubMed] [CrossRef]
161. Ray A, Kunhiraman H, Perera RJ. The paradoxical behavior of microRNA-211 in melanomas and other human cancers. Front Oncol. 2021;10:628367. doi:10.3389/fonc.2020.628367. [Google Scholar] [PubMed] [CrossRef]
162. Ostrowski SM, Fisher DE. The melanocyte lineage factor miR-211 promotes BRAFV600E inhibitor resistance. J Invest Dermatol. 2021;141(2):250–2. doi:10.1016/j.jid.2020.07.010. [Google Scholar] [PubMed] [CrossRef]
163. Zhang J, Liu G, Jin H, Li X, Li N, Yin Q, et al. MicroRNA-137 targets EZH2 to exert suppressive functions in uveal melanoma via regulation of Wnt/β-catenin signaling and epithelial-to-mesenchymal transition. J BUON. 2021;26(1):173–81. [Google Scholar] [PubMed]
164. Li WJ, Wang Y, Liu R, Kasinski AL, Shen H, Slack FJ, et al. MicroRNA-34a: potent tumor suppressor, cancer stem cell inhibitor, and potential anticancer therapeutic. Front Cell Dev Biol. 2021;9:640587. doi:10.3389/fcell.2021.640587. [Google Scholar] [PubMed] [CrossRef]
165. Mazar J, Khaitan D, DeBlasio D, Zhong C, Govindarajan SS, Kopanathi S, et al. Epigenetic regulation of microRNA genes and the role of miR-34b in cell invasion and motility in human melanoma. PLoS One. 2011;6(9):e24922. doi:10.1371/journal.pone.0024922. [Google Scholar] [PubMed] [CrossRef]
166. Vera O, Bok I, Jasani N, Nakamura K, Xu X, Mecozzi N, et al. A MAPK/miR-29 axis suppresses melanoma by targeting MAFG and MYBL2. Cancers. 2021;13(6):1408. doi:10.3390/cancers13061408. [Google Scholar] [PubMed] [CrossRef]
167. Tang H, Ma M, Dai J, Cui C, Si L, Sheng X, et al. miR-let-7b and miR-let-7c suppress tumourigenesis of human mucosal melanoma and enhance the sensitivity to chemotherapy. J Exp Clin Cancer Res. 2019;38(1):212. doi:10.1186/s13046-019-1190-3. [Google Scholar] [PubMed] [CrossRef]
168. Gao Y, Xu J, Li H, Hu Y, Yu G. Identification of metastasis-associated MicroRNAs in metastatic melanoma by miRNA expression profile and experimental validation. Front Genet. 2021;12:663110. doi:10.3389/fgene.2021.663110. [Google Scholar] [PubMed] [CrossRef]
169. Yin Y, Cai X, Chen X, Liang H, Zhang Y, Li J, et al. Tumor-secreted miR-214 induces regulatory T cells: a major link between immune evasion and tumor growth. Cell Res. 2014;24(10):1164–80. doi:10.1038/cr.2014.121. [Google Scholar] [PubMed] [CrossRef]
170. Fattore L, Costantini S, Malpicci D, Ruggiero CF, Ascierto PA, Croce CM, et al. MicroRNAs in melanoma development and resistance to target therapy. Oncotarget. 2017;8(13):22262–78. doi:10.18632/oncotarget.14763. [Google Scholar] [PubMed] [CrossRef]
171. Trujillo JA, Luke JJ, Zha Y, Segal JP, Ritterhouse LL, Spranger S, et al. Secondary resistance to immunotherapy associated with β-catenin pathway activation or PTEN loss in metastatic melanoma. J Immunother Cancer. 2019;7(1):295. doi:10.1186/s40425-019-0780-0. [Google Scholar] [PubMed] [CrossRef]
172. Kewitz-Hempel S, Windisch N, Hause G, Müller L, Sunderkötter C, Gerloff D. Extracellular vesicles derived from melanoma cells induce carcinoma-associated fibroblasts via miR-92b-3p mediated downregulation of PTEN. J Extracell Vesicles. 2024;13(9):e12509. doi:10.1002/jev2.12509. [Google Scholar] [PubMed] [CrossRef]
173. Haflidadóttir BS, Bergsteinsdóttir K, Praetorius C, Steingrímsson E. miR-148 regulates Mitf in melanoma cells. PLoS One. 2010;5(7):e11574. doi:10.1371/journal.pone.0011574. [Google Scholar] [PubMed] [CrossRef]
174. Luo C, Tetteh PW, Merz PR, Dickes E, Abukiwan A, Hotz-Wagenblatt A, et al. miR-137 inhibits the invasion of melanoma cells through downregulation of multiple oncogenic target genes. J Invest Dermatol. 2013;133(3):768–75. doi:10.1038/jid.2012.357. [Google Scholar] [PubMed] [CrossRef]
175. Luo C, Merz PR, Chen Y, Dickes E, Pscherer A, Schadendorf D, et al. MiR-101 inhibits melanoma cell invasion and proliferation by targeting MITF and EZH2. Cancer Lett. 2013;341(2):240–7. doi:10.1016/j.canlet.2013.08.021. [Google Scholar] [PubMed] [CrossRef]
176. Liguoro D, Frigerio R, Ortolano A, Sacconi A, Acunzo M, Romano G, et al. The MITF/mir-579-3p regulatory axis dictates BRAF-mutated melanoma cell fate in response to MAPK inhibitors. Cell Death Dis. 2024;15(3):208. doi:10.1038/s41419-024-06580-2. [Google Scholar] [PubMed] [CrossRef]
177. Lai X, Luan C, Zhang Z, Wessely A, Heppt MV, Berking C, et al. SOX10, MITF, and microRNAs: decoding their interplay in regulating melanoma plasticity. Int J Cancer. 2025;157(7):1277–93. doi:10.1002/ijc.35499. [Google Scholar] [PubMed] [CrossRef]
178. Castellani G, Buccarelli M, Arasi MB, Rossi S, Pisanu ME, Bellenghi M, et al. BRAF Mutations in melanoma: biological aspects, therapeutic implications, and circulating biomarkers. Cancers. 2023;15(16):4026. doi:10.3390/cancers15164026. [Google Scholar] [PubMed] [CrossRef]
179. Varrone F, Caputo E. The miRNAs role in melanoma and in its resistance to therapy. Int J Mol Sci. 2020;21(3):878. doi:10.3390/ijms21030878. [Google Scholar] [PubMed] [CrossRef]
180. Motti ML, Minopoli M, Di Carluccio G, Ascierto PA, Carriero MV. MicroRNAs as key players in melanoma cell resistance to MAPK and immune checkpoint inhibitors. Int J Mol Sci. 2020;21(12):4544. doi:10.3390/ijms21124544. [Google Scholar] [PubMed] [CrossRef]
181. Afsar S, Syed RU, Khojali WMA, Masood N, Osman ME, Jyothi JS, et al. Non-coding RNAs in BRAF-mutant melanoma: targets, indicators, and therapeutic potential. Naunyn Schmiedebergs Arch Pharmacol. 2025;398(1):297–317. doi:10.1007/s00210-024-03366-3. [Google Scholar] [PubMed] [CrossRef]
182. Fattore L, Mancini R, Acunzo M, Romano G, Laganà A, Pisanu ME, et al. miR-579-3p controls melanoma progression and resistance to target therapy. Proc Natl Acad Sci U S A. 2016;113(34):E5005–13. doi:10.1073/pnas.1607753113. [Google Scholar] [PubMed] [CrossRef]
183. Loureiro JB, Raimundo L, Calheiros J, Carvalho C, Barcherini V, Lima NR, et al. Targeting p53 for melanoma treatment: counteracting tumour proliferation, dissemination and therapeutic resistance. Cancers. 2021;13(7):1648. doi:10.3390/cancers13071648. [Google Scholar] [PubMed] [CrossRef]
184. Ingelshed K, Spiegelberg D, Kannan P, Påvénius L, Hacheney J, Jiang L, et al. The MDM2 inhibitor navtemadlin arrests mouse melanoma growth in vivo and potentiates radiotherapy. Cancer Res Commun. 2022;2(9):1075–88. doi:10.1158/2767-9764.CRC-22-0053. [Google Scholar] [PubMed] [CrossRef]
185. Wang W, Albadari N, Du Y, Fowler JF, Sang HT, Xian W, et al. MDM2 inhibitors for cancer therapy: the past, present, and future. Pharmacol Rev. 2024;76(3):414–53. doi:10.1124/pharmrev.123.001026. [Google Scholar] [PubMed] [CrossRef]
186. Zhang W, Mao K, Liu S, Xu Y, Ren J. miR-942-5p promotes the proliferation and invasion of human melanoma cells by targeting DKK3. J Recept Signal Transduct Res. 2021;41(2):180–7. doi:10.1080/10799893.2020.1804280. [Google Scholar] [PubMed] [CrossRef]
187. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3(6):401–10. doi:10.1038/nrc1093. [Google Scholar] [PubMed] [CrossRef]
188. Girouard SD, Murphy GF. Melanoma stem cells: not rare, but well done. Lab Invest. 2011;91(5):647–64. doi:10.1038/labinvest.2011.50. [Google Scholar] [PubMed] [CrossRef]
189. Tupone MG, D’Aguanno S, Di Martile M, Valentini E, Desideri M, Trisciuoglio D, et al. microRNA-378a-5p iS a novel positive regulator of melanoma progression. Oncogenesis. 2020;9(2):22. doi:10.1038/s41389-020-0203-6. [Google Scholar] [PubMed] [CrossRef]
190. Zhou X, Yan T, Huang C, Xu Z, Wang L, Jiang E, et al. Melanoma cell-secreted exosomal miR-155-5p induce proangiogenic switch of cancer-associated fibroblasts via SOCS1/JAK2/STAT3 signaling pathway. J Exp Clin Cancer Res. 2018;37(1):242. doi:10.1186/s13046-018-0911-3. [Google Scholar] [PubMed] [CrossRef]
191. Yu H, Yang W. MiR-211 is epigenetically regulated by DNMT1 mediated methylation and inhibits EMT of melanoma cells by targeting RAB22A. Biochem Biophys Res Commun. 2016;476(4):400–5. doi:10.1016/j.bbrc.2016.05.133. [Google Scholar] [PubMed] [CrossRef]
192. Tigu AB, Ivancuta A, Uhl A, Sabo AC, Nistor M, Muresan XM, et al. Epigenetic therapies in melanoma-targeting DNA methylation and histone modification. Biomedicines. 2025;13(5):1188. doi:10.3390/biomedicines13051188. [Google Scholar] [PubMed] [CrossRef]
193. Wozniak M, Sztiller-Sikorska M, Czyz M. Diminution of miR-340-5p levels is responsible for increased expression of ABCB5 in melanoma cells under oxygen-deprived conditions. Exp Mol Pathol. 2015;99(3):707–16. doi:10.1016/j.yexmp.2015.11.014. [Google Scholar] [PubMed] [CrossRef]
194. Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, et al. Identification of cells initiating human melanomas. Nature. 2008;451(7176):345–9. doi:10.1038/nature06489. [Google Scholar] [PubMed] [CrossRef]
195. Wilson BJ, Saab KR, Ma J, Schatton T, Pütz P, Zhan Q, et al. ABCB5 maintains melanoma-initiating cells through a proinflammatory cytokine signaling circuit. Cancer Res. 2014;74(15):4196–207. doi:10.1158/0008-5472.CAN-14-0582. [Google Scholar] [PubMed] [CrossRef]
196. Wanior M, Krämer A, Knapp S, Joerger AC. Exploiting vulnerabilities of SWI/SNF chromatin remodelling complexes for cancer therapy. Oncogene. 2021;40(21):3637–54. doi:10.1038/s41388-021-01781-x. [Google Scholar] [PubMed] [CrossRef]
197. Innis SM, Cabot B. GBAF, a small BAF sub-complex with big implications: a systematic review. Epigenetics Chromatin. 2020;13(1):48. doi:10.1186/s13072-020-00370-8. [Google Scholar] [PubMed] [CrossRef]
198. Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One. 2013;8(1):e55119. doi:10.1371/journal.pone.0055119. [Google Scholar] [PubMed] [CrossRef]
199. Saladi SV, Keenen B, Marathe HG, Qi H, Chin KV, de la Serna IL. Modulation of extracellular matrix/adhesion molecule expression by BRG1 is associated with increased melanoma invasiveness. Mol Cancer. 2010;9:280. doi:10.1186/1476-4598-9-280. [Google Scholar] [PubMed] [CrossRef]
200. Laurette P, Coassolo S, Davidson G, Michel I, Gambi G, Yao W, et al. Chromatin remodellers Brg1 and Bptf are required for normal gene expression and progression of oncogenic Braf-driven mouse melanoma. Cell Death Differ. 2020;27(1):29–43. doi:10.1038/s41418-019-0333-6. [Google Scholar] [PubMed] [CrossRef]
201. Rago F, Elliott G, Li A, Sprouffske K, Kerr G, Desplat A, et al. The discovery of SWI/SNF chromatin remodeling activity as a novel and targetable dependency in uveal melanoma. Mol Cancer Ther. 2020;19(10):2186–95. doi:10.1158/1535-7163.MCT-19-1013. [Google Scholar] [PubMed] [CrossRef]
202. Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132(3):363–74. doi:10.1016/j.cell.2007.12.032. [Google Scholar] [PubMed] [CrossRef]
203. Aras S, Saladi SV, Basuroy T, Marathe HG, Lorès P, de la Serna IL. BAF60A mediates interactions between the microphthalmia-associated transcription factor and the BRG1-containing SWI/SNF complex during melanocyte differentiation. J Cell Physiol. 2019;234(7):11780–91. doi:10.1002/jcp.27840. [Google Scholar] [PubMed] [CrossRef]
204. Zhao DD, Zhao X, Li WT. Identification of differentially expressed metastatic genes and their signatures to predict the overall survival of uveal melanoma patients by bioinformatics analysis. Int J Ophthalmol. 2020;13(7):1046–53. doi:10.18240/ijo.2020.07.05. [Google Scholar] [PubMed] [CrossRef]
205. Mendiratta G, Ke E, Aziz M, Liarakos D, Tong M, Stites EC. Cancer gene mutation frequencies for the U.S. population. Nat Commun. 2021;12(1):5961. doi:10.1038/s41467-021-26213-y. [Google Scholar] [PubMed] [CrossRef]
206. Li J, Wang W, Zhang Y, Cieślik M, Guo J, Tan M, et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J Clin Invest. 2020;130(5):2712–26. doi:10.1172/JCI134402. [Google Scholar] [PubMed] [CrossRef]
207. Broit N, Johansson PA, Rodgers CB, Walpole ST, Newell F, Hayward NK, et al. Meta-analysis and systematic review of the genomics of mucosal melanoma. Mol Cancer Res. 2021;19(6):991–1004. doi:10.1158/1541-7786.MCR-20-0839. [Google Scholar] [PubMed] [CrossRef]
208. Shain AH, Joseph NM, Yu R, Benhamida J, Liu S, Prow T, et al. Genomic and transcriptomic analysis reveals incremental disruption of key signaling pathways during melanoma evolution. Cancer Cell. 2018;34(1):45–55.e4. doi:10.1016/j.ccell.2018.06.005. [Google Scholar] [PubMed] [CrossRef]
209. Fukumoto T, Lin J, Fatkhutdinov N, Liu P, Somasundaram R, Herlyn M, et al. ARID2 deficiency correlates with the response to immune checkpoint blockade in melanoma. J Invest Dermatol. 2021;141(6):1564–72.e4. doi:10.1016/j.jid.2020.11.026. [Google Scholar] [PubMed] [CrossRef]
210. Soshnikova NV, Tatarskiy EV, Tatarskiy VV, Klimenko NS, Shtil AA, Nikiforov MA, et al. PHF10 subunit of PBAF complex mediates transcriptional activation by MYC. Oncogene. 2021;40(42):6071–80. doi:10.1038/s41388-021-01994-0. [Google Scholar] [PubMed] [CrossRef]
211. Mason LD, Chava S, Reddi KK, Gupta R. The BRD9/7 inhibitor TP-472 blocks melanoma tumor growth by suppressing ECM-mediated oncogenic signaling and inducing apoptosis. Cancers. 2021;13(21):5516. doi:10.3390/cancers13215516. [Google Scholar] [PubMed] [CrossRef]
212. Dar AA, Nosrati M, Bezrookove V, de Semir D, Majid S, Thummala S, et al. The role of BPTF in melanoma progression and in response to BRAF-targeted therapy. J Natl Cancer Inst. 2015;107(5):djv034. doi:10.1093/jnci/djv034. [Google Scholar] [PubMed] [CrossRef]
213. Qadeer ZA, Harcharik S, Valle-Garcia D, Chen C, Birge MB, Vardabasso C, et al. Decreased expression of the chromatin remodeler ATRX associates with melanoma progression. J Invest Dermatol. 2014;134(6):1768–72. doi:10.1038/jid.2014.45. [Google Scholar] [PubMed] [CrossRef]
214. Lally SE, Milman T, Orloff M, Dalvin LA, Eberhart CG, Heaphy CM, et al. Mutational landscape and outcomes of conjunctival melanoma in 101 patients. Ophthalmology. 2022;129(6):679–93. doi:10.1016/j.ophtha.2022.01.016. [Google Scholar] [PubMed] [CrossRef]
215. Van Ipenburg JA, Van den Bosch QCC, Paridaens D, Dubbink HJ, Kiliç E, Naus N, et al. Rotterdam ocular melanoma study group. ATRX loss in the development and prognosis of conjunctival melanoma. Int J Mol Sci. 2023;24(16):12988. doi:10.3390/ijms241612988. [Google Scholar] [PubMed] [CrossRef]
216. Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169(7):1187–200. doi:10.1016/j.cell.2017.05.045. [Google Scholar] [PubMed] [CrossRef]
217. Machnicka MA, Olchowik A, Grosjean H, Bujnicki JM. Distribution and frequencies of post-transcriptional modifications in tRNAs. RNA Biol. 2014;11(12):1619–29. doi:10.4161/15476286.2014.992273. [Google Scholar] [PubMed] [CrossRef]
218. Decatur WA, Fournier MJ. rRNA modifications and ribosome function. Trends Biochem Sci. 2002;27(7):344–51. doi:10.1016/s0968-0004(02)02109-6. [Google Scholar] [PubMed] [CrossRef]
219. Boo SH, Kim YK. The emerging role of RNA modifications in the regulation of mRNA stability. Exp Mol Med. 2020;52(3):400–8. doi:10.1038/s12276-020-0407-z. [Google Scholar] [PubMed] [CrossRef]
220. Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. 2014;15(5):293–306. doi:10.1038/nrg3724. [Google Scholar] [PubMed] [CrossRef]
221. Qu L, Liu SJ, Zhang L, Liu JF, Zhou YJ, Zeng PH, et al. The role of m6A-mediated DNA damage repair in tumor development and chemoradiotherapy resistance. Cancer Control. 2024;31:10732748241247170. doi:10.1177/10732748241247170. [Google Scholar] [PubMed] [CrossRef]
222. Liao J, Wei Y, Liang J, Wen J, Chen X, Zhang B, et al. Insight into the structure, physiological function, and role in cancer of m6A readers-YTH domain-containing proteins. Cell Death Discov. 2022;8(1):137. doi:10.1038/s41420-022-00947-0. [Google Scholar] [PubMed] [CrossRef]
223. Wang G, Zeng D, Sweren E, Miao Y, Chen R, Chen J, et al. m6A RNA methylation correlates with immune microenvironment and immunotherapy response of melanoma. J Invest Dermatol. 2023;143(8):1579–90.e5. doi:10.1016/j.jid.2023.01.027. [Google Scholar] [PubMed] [CrossRef]
224. Malvi P, Wang B, Shah S, Gupta R. Dissecting the role of RNA modification regulatory proteins in melanoma. Oncotarget. 2019;10(38):3745–59. doi:10.18632/oncotarget.26959. [Google Scholar] [CrossRef]
225. Wu H, Xu H, Jia D, Li T, Xia L. METTL3-induced UCK2 m6A hypermethylation promotes melanoma cancer cell metastasis via the WNT/β-catenin pathway. Ann Transl Med. 2021;9(14):1155. doi:10.21037/atm-21-2906. [Google Scholar] [PubMed] [CrossRef]
226. Wu X, Chen D, Li M, Liang G, Ye H. UCK2 promotes intrahepatic cholangiocarcinoma progression and desensitizes cisplatin treatment by PI3K/AKT/mTOR/autophagic axis. Cell Death Discov. 2024;10(1):375. doi:10.1038/s41420-024-02140-x. [Google Scholar] [PubMed] [CrossRef]
227. Liu S, Zhuo L, Wang J, Zhang Q, Li Q, Li G, et al. METTL3 plays multiple functions in biological processes. Am J Cancer Res. 2020;10(6):1631–46. [Google Scholar] [PubMed]
228. Dahal U, Le K, Gupta M. RNA m6A methyltransferase METTL3 regulates invasiveness of melanoma cells by matrix metallopeptidase 2. Melanoma Res. 2019;29(4):382–9. doi:10.1097/CMR.0000000000000580. [Google Scholar] [PubMed] [CrossRef]
229. Li N, Kang Y, Wang L, Huff S, Tang R, Hui H, et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci U S A. 2020;117(33):20159–70. doi:10.1073/pnas.1918986117. [Google Scholar] [PubMed] [CrossRef]
230. Hao L, Yin J, Yang H, Li C, Zhu L, Liu L, et al. ALKBH5-mediated m6A demethylation of FOXM1 mRNA promotes progression of uveal melanoma. Aging. 2021;13(3):4045–62. doi:10.18632/aging.202371. [Google Scholar] [PubMed] [CrossRef]
231. Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, et al. m6A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10(1):2782. doi:10.1038/s41467-019-10669-0. [Google Scholar] [PubMed] [CrossRef]
232. Zhang Y, Li L, Mendoza JJ, Wang D, Yan Q, Shi L, et al. Advances in A-to-I RNA editing in cancer. Mol Cancer. 2024;23(1):280. doi:10.1186/s12943-024-02194-6. [Google Scholar] [PubMed] [CrossRef]
233. Siddiqui J, Miles WO. RNA editing signatures identify melanoma patients who respond to Pembrolizumab or Nivolumab treatment. Transl Oncol. 2021;14(11):101197. doi:10.1016/j.tranon.2021.101197. [Google Scholar] [PubMed] [CrossRef]
234. Vlachogiannis NI, Polycarpou-Schwarz M, Avdi AP, Tual-Chalot S, Stellos K. Targeting RNA adenosine editing and modification enzymes for RNA therapeutics. Mol Ther. 2025;33(9):4044–90. doi:10.1016/j.ymthe.2025.05.021. [Google Scholar] [PubMed] [CrossRef]
235. Chen YP, Hou XY, Yang CS, Jiang XX, Yang M, Xu XF, et al. DNA methylation and histone acetylation regulate the expression of MGMT and chemosensitivity to temozolomide in malignant melanoma cell lines. Tumour Biol. 2016;37(8):11209–18. doi:10.1007/s13277-016-4994-1. [Google Scholar] [PubMed] [CrossRef]
236. Hassel JC, Sucker A, Edler L, Kurzen H, Moll I, Stresemann C, et al. MGMT gene promoter methylation correlates with tolerance of temozolomide treatment in melanoma but not with clinical outcome. Br J Cancer. 2010;103(6):820–6. doi:10.1038/sj.bjc.6605796. [Google Scholar] [PubMed] [CrossRef]
237. Guadagni S, Fiorentini G, Clementi M, Palumbo G, Masedu F, Deraco M, et al. MGMT methylation correlateswith melphalan pelvic perfusion survival in stage III melanoma patients: a pilot study. Melanoma Res. 2017;27(5):439–447. doi:10.1097/CMR.0000000000000367. [Google Scholar] [PubMed] [CrossRef]
238. Tellez CS, Shen L, Estécio MR, Jelinek J, Gershenwald JE, Issa JP. CpG island methylation profiling in human melanoma cell lines. Melanoma Res. 2009;19(3):146–55. doi:10.1097/cmr.0b013e32832b274e. [Google Scholar] [PubMed] [CrossRef]
239. Tanemura A, Terando AM, Sim MS, Van Hoesel AQ, de Maat MF, Morton DL, et al. CpG island methylator phenotype predicts progression of malignant melanoma. Clin Cancer Res. 2009;15(5):1801–7. doi:10.1158/1078-0432.CCR-08-1361. [Google Scholar] [PubMed] [CrossRef]
240. Shao C, Dai W, Li H, Tang W, Jia S, Wu X, et al. The relationship between RASSF1A gene promoter methylation and the susceptibility and prognosis of melanoma: a meta-analysis and bioinformatics. PLoS One. 2017;12(2):e0171676. doi:10.1371/journal.pone.0171676. [Google Scholar] [PubMed] [CrossRef]
241. Salvianti F, Orlando C, Massi D, De Giorgi V, Grazzini M, Pazzagli M, et al. Tumor-related methylated cell-free DNA and circulating tumor cells in melanoma. Front Mol Biosci. 2016;2:76. doi:10.3389/fmolb.2015.00076. [Google Scholar] [PubMed] [CrossRef]
242. Jonsson A, Tuominen R, Grafström E, Hansson J, Egyhazi S. High frequency of p16INK4A promoter methylation in NRAS-mutated cutaneous melanoma. J Invest Dermatol. 2010;130(12):2809–17. doi:10.1038/jid.2010.216. [Google Scholar] [PubMed] [CrossRef]
243. Straume O, Sviland L, Akslen LA. Loss of nuclear p16 protein expression correlates with increased tumor cell proliferation (Ki-67) and poor prognosis in patients with vertical growth phase melanoma. Clin Cancer Res. 2000;6(5):1845–53. [Google Scholar] [PubMed]
244. Conway K, Tsai YS, Edmiston SN, Parker JS, Parrish EA, Hao H, et al. Characterization of the CpG Island hypermethylated phenotype subclass in primary melanomas. J Invest Dermatol. 2022;142(7):1869–1881.e10. doi:10.1016/j.jid.2021.11.017. [Google Scholar] [PubMed] [CrossRef]
245. Micevic G, Thakral D, McGeary M, Bosenberg MW. PD-L1 methylation regulates PD-L1 expression and is associated with melanoma survival. Pigment Cell Melanoma Res. 2019;32(3):435–40. doi:10.1111/pcmr.12745. [Google Scholar] [PubMed] [CrossRef]
246. Goltz D, Gevensleben H, Vogt TJ, Dietrich J, Golletz C, Bootz F, et al. CTLA4 methylation predicts response to anti-PD-1 and anti-CTLA-4 immunotherapy in melanoma patients. JCI Insight. 2018;3(13):e96793. doi:10.1172/jci.insight.96793. [Google Scholar] [PubMed] [CrossRef]
247. Roh MR, Gupta S, Park KH, Chung KY, Lauss M, Flaherty KT, et al. Promoter methylation of PTEN is a significant prognostic factor in melanoma survival. J Invest Dermatol. 2016;136(5):1002–11. doi:10.1016/j.jid.2016.01.024. [Google Scholar] [PubMed] [CrossRef]
248. Marzese DM, Scolyer RA, Huynh JL, Huang SK, Hirose H, Chong KK, et al. Epigenome-wide DNA methylation landscape of melanoma progression to brain metastasis reveals aberrations on homeobox D cluster associated with prognosis. Hum Mol Genet. 2014;23(1):226–38. doi:10.1093/hmg/ddt420. [Google Scholar] [PubMed] [CrossRef]
249. Muralidhar S, Filia A, Nsengimana J, Poźniak J, O’Shea SJ, Diaz JM, et al. Vitamin D-VDR signaling inhibits Wnt/β-catenin-mediated melanoma progression and promotes antitumor immunity. Cancer Res. 2019;79(23):5986–98. doi:10.1158/0008-5472.CAN-18-3927. [Google Scholar] [PubMed] [CrossRef]
250. Sigalotti L, Covre A, Fratta E, Parisi G, Sonego P, Colizzi F, et al. Whole genome methylation profiles as independent markers of survival in stage IIIC melanoma patients. J Transl Med. 2012;10:185. doi:10.1186/1479-5876-10-185. [Google Scholar] [PubMed] [CrossRef]
251. Qi F, Yin Z, Wang G, Zeng S. Clinical and prognostic significance of O6-methylguanine-DNA methyltransferase promoter methylation in patients with melanoma: a systematic meta-analysis. Ann Dermatol. 2018;30(2):129–35. doi:10.5021/ad.2018.30.2.129. [Google Scholar] [PubMed] [CrossRef]
252. Guo W, Zhu L, Zhu R, Chen Q, Wang Q, Chen JQ. A four-DNA methylation biomarker is a superior predictor of survival of patients with cutaneous melanoma. eLife. 2019;8:e44310. doi:10.7554/eLife.44310. [Google Scholar] [PubMed] [CrossRef]
253. Pradhan D, Jour G, Milton D, Vasudevaraja V, Tetzlaff MT, Nagarajan P, et al. Aberrant DNA methylation predicts melanoma-specific survival in patients with acral melanoma. Cancers. 2019;11(12):2031. doi:10.3390/cancers11122031. [Google Scholar] [PubMed] [CrossRef]
254. Yang J, Dong M, Shui Y, Zhang Y, Zhang Z, Mi Y, et al. A pooled analysis of the prognostic value of PD-L1 in melanoma: evidence from 1062 patients. Cancer Cell Int. 2020;20:96. doi:10.1186/s12935-020-01187-x. [Google Scholar] [PubMed] [CrossRef]
255. Rius FE, Papaiz DD, Azevedo HFZ, Ayub ALP, Pessoa DO, Oliveira TF, et al. Genome-wide promoter methylation profiling in a cellular model of melanoma progression reveals markers of malignancy and metastasis that predict melanoma survival. Clin Epigenetics. 2022;14(1):68. doi:10.1186/s13148-022-01291-x. [Google Scholar] [PubMed] [CrossRef]
256. Rodić N, Zampella J, Sharma R, Burns KH, Taube JM. Diagnostic utility of 5-hydroxymethylcytosine immunohistochemistry in melanocytic proliferations. J Cutan Pathol. 2015;42(11):807–14. doi:10.1111/cup.12564. [Google Scholar] [PubMed] [CrossRef]
257. Fischer AP, Miles SL. Silencing HIF-1α induces TET2 expression and augments ascorbic acid induced 5-hydroxymethylation of DNA in human metastatic melanoma cells. Biochem Biophys Res Commun. 2017;490(2):176–81. doi:10.1016/j.bbrc.2017.06.017. [Google Scholar] [PubMed] [CrossRef]
258. Yu Y, Hosseini N, Dodington D, Wood K, Ghazarian D, Kamil ZS. The combined diagnostic value of 5-hmC and PRAME immunohistochemistry in melanocytic neoplasms. Pathol Res Pract. 2025;270:155993. doi:10.1016/j.prp.2025.155993. [Google Scholar] [PubMed] [CrossRef]
259. Stahl A, Riggi N, Nardou K, Nicolas M, Kaya G, Moulin A. 5-hydroxymethylcytosine loss in conjunctival melanoma. Dermatopathology. 2021;8(2):176–84. doi:10.3390/dermatopathology8020023. [Google Scholar] [PubMed] [CrossRef]
260. Salgado C, Oosting J, Janssen B, Kumar R, Gruis N, Van Doorn R. Genome-wide characterization of 5-hydoxymethylcytosine in melanoma reveals major differences with nevus. Genes Chromosomes Cancer. 2020;59(6):366–74. doi:10.1002/gcc.22837. [Google Scholar] [PubMed] [CrossRef]
261. Gomes CB, Zechin KG, Xu S, Stelini RF, Nishimoto IN, Zhan Q, et al. TET2 negatively regulates nestin expression in human melanoma. Am J Pathol. 2016;186(6):1427–34. doi:10.1016/j.ajpath.2016.01.020. [Google Scholar] [PubMed] [CrossRef]
262. Lee JJ, Vilain RE, Granter SR, Hu NR, Bresler SC, Xu S, et al. 5-Hydroxymethylcytosine is a nuclear biomarker to assess biological potential in histologically ambiguous heavily pigmented melanocytic neoplasms. J Cutan Pathol. 2017;44(3):249–55. doi:10.1111/cup.12880. [Google Scholar] [PubMed] [CrossRef]
263. Saldanha G, Joshi K, Lawes K, Bamford M, Moosa F, Teo KW, et al. 5-Hydroxymethylcytosine is an independent predictor of survival in malignant melanoma. Mod Pathol. 2017;30(1):60–8. doi:10.1038/modpathol.2016.159. [Google Scholar] [PubMed] [CrossRef]
264. Kuźbicki Ł. The markers auxiliary in distinguishing between nodal nevi and melanoma metastases. Curr Med Chem. 2025;32:8580–97. doi:10.2174/0109298673358592250408170859. [Google Scholar] [PubMed] [CrossRef]
265. Katarzyna L, Kyriakos O, Linda V, Ingrid S, Petra W, Karin Ö. Evaluation of tubulin β-3 and 5 hydroxy-methyl cytosine as diagnostic and prognostic markers in malignant melanoma. Ann Diagn Pathol. 2024;72:152332. doi:10.1016/j.anndiagpath.2024.152332. [Google Scholar] [PubMed] [CrossRef]
266. Koch EAT, Berking C, Erber R, Erdmann M, Kiesewetter F, Schliep S, et al. Standardized computer-assisted analysis of 5-hmC immunoreactivity in dysplastic nevi and superficial spreading melanomas. Int J Mol Sci. 2023;24(19):14711. doi:10.3390/ijms241914711. [Google Scholar] [PubMed] [CrossRef]
267. Lin Z, Yang L. Identification of a CpG-based signature coupled with gene expression as prognostic indicators for melanoma: a preliminary study. Sci Rep. 2024;14(1):5302. doi:10.1038/s41598-023-50614-2. [Google Scholar] [PubMed] [CrossRef]
268. Rawson RV, Shteinman ER, Ansar S, Vergara IA, Thompson JF, Long GV, et al. Diagnostic utility of PRAME, p53 and 5-hmC immunostaining for distinguishing melanomas from naevi, neurofibromas, scars and other histological mimics. Pathology. 2022;54(7):863–73. doi:10.1016/j.pathol.2022.05.012. [Google Scholar] [PubMed] [CrossRef]
269. Li FJ, Li LM, Zhang RH, Xu C, Zhou P, Long J, et al. The role of 5-hydroxymethylcytosine in melanoma. Melanoma Res. 2017;27(3):175–9. doi:10.1097/CMR.0000000000000349. [Google Scholar] [PubMed] [CrossRef]
270. Wu N, Sun H, Sun Q, Zhang F, Ma L, Hu Y, et al. Circulating microRNAs as diagnostic biomarkers for melanoma: a systematic review and meta-analysis. BMC Cancer. 2023;23(1):414. doi:10.1186/s12885-023-10891-6. PMID: 37158840; PMCID: PMC10165832. [Google Scholar] [PubMed] [CrossRef]
271. Van Laar R, Lincoln M, Van Laar B. Development and validation of a plasma-based melanoma biomarker suitable for clinical use. Br J Cancer. 2018;118(6):857–66. doi:10.1038/bjc.2017.477. [Google Scholar] [PubMed] [CrossRef]
272. Love CG, Coombs L, Van Laar R. RNA-seq validation of microRNA expression signatures for precision melanoma diagnosis and prognostic stratification. BMC Med Genomics. 2024;17(1):256. doi:10.1186/s12920-024-02028-w. [Google Scholar] [PubMed] [CrossRef]
273. Sabato C, Noviello TMR, Covre A, Coral S, Caruso FP, Besharat ZM, et al. A novel microRNA signature for the detection of melanoma by liquid biopsy. J Transl Med. 2022;20(1):469. doi:10.1186/s12967-022-03668-1. [Google Scholar] [PubMed] [CrossRef]
274. Jones N, Nonaka T. Circulating miRNAs as biomarkers for the diagnosis in patients with melanoma: systematic review and meta-analysis. Front Genet. 2024;15:1339357. doi:10.3389/fgene.2024.1339357. [Google Scholar] [PubMed] [CrossRef]
275. Friedman EB, Shang S, de Miera EV, Fog JU, Teilum MW, Ma MW, et al. Serum microRNAs as biomarkers for recurrence in melanoma. J Transl Med. 2012;10:155. doi:10.1186/1479-5876-10-155. [Google Scholar] [PubMed] [CrossRef]
276. Stark MS, Klein K, Weide B, Haydu LE, Pflugfelder A, Tang YH, et al. The prognostic and predictive value of melanoma-related MicroRNAs using tissue and serum: a microRNA expression analysis. eBioMedicine. 2015;2(7):671–80. doi:10.1016/j.ebiom.2015.05.011. [Google Scholar] [PubMed] [CrossRef]
277. Fleming NH, Zhong J, da Silva IP, Vega-Saenz de Miera E, Brady B, Han SW, et al. Serum-based miRNAs in the prediction and detection of recurrence in melanoma patients. Cancer. 2015;121(1):51–9. doi:10.1002/cncr.28981. [Google Scholar] [PubMed] [CrossRef]
278. Bustos MA, Tran KD, Rahimzadeh N, Gross R, Lin SY, Shoji Y, et al. Integrated assessment of circulating cell-free MicroRNA signatures in plasma of patients with melanoma brain metastasis. Cancers. 2020;12(6):1692. doi:10.3390/cancers12061692. [Google Scholar] [PubMed] [CrossRef]
279. Borea R, Saldanha EF, Maheswaran S, Nicolo E, Singhal S, Pontolillo L, et al. Cancer in a drop: advances in liquid biopsy in 2024. Crit Rev Oncol Hematol. 2025;213:104776. doi:10.1016/j.critrevonc.2025.104776. [Google Scholar] [PubMed] [CrossRef]
280. Slusher N, Jones N, Nonaka T. Liquid biopsy for diagnostic and prognostic evaluation of melanoma. Front Cell Dev Biol. 2024;12:1420360. doi:10.3389/fcell.2024.1420360. [Google Scholar] [PubMed] [CrossRef]
281. Chen Z, Li C, Zhou Y, Yao Y, Liu J, Wu M, et al. Liquid biopsies for cancer: from bench to clinic. MedComm. 2023;4(4):e329. doi:10.1002/mco2.329. [Google Scholar] [PubMed] [CrossRef]
282. Schroeder C, Gatidis S, Kelemen O, Schütz L, Bonzheim I, Muyas F, et al. Tumour-informed liquid biopsies to monitor advanced melanoma patients under immune checkpoint inhibition. Nat Commun. 2024;15(1):8750. doi:10.1038/s41467-024-52923-0. [Google Scholar] [PubMed] [CrossRef]
283. Genta S, Araujo DV, Hueniken K, Pipinikas C, Ventura R, Rojas P, et al. Bespoke ctDNA for longitudinal detection of molecular residual disease in high-risk melanoma patients. ESMO Open. 2024;9(11):103978. doi:10.1016/j.esmoop.2024.103978. [Google Scholar] [PubMed] [CrossRef]
284. Kim YJ, Rho WY, Park SM, Jun BH. Optical nanomaterial-based detection of biomarkers in liquid biopsy. J Hematol Oncol. 2024;17(1):10. doi:10.1186/s13045-024-01531-y. [Google Scholar] [PubMed] [CrossRef]
285. Noreen S, Ishaq I, Saleem MH, Ali B, Muhammad Ali S, Iqbal J. Electrochemical biosensing in oncology: a review advancements and prospects for cancer diagnosis. Cancer Biol Ther. 2025;26(1):2475581. doi:10.1080/15384047.2025.2475581. [Google Scholar] [PubMed] [CrossRef]
286. Tolu SS, Viny AD, Amengual JE, Pro B, Bates SE. Getting the right combination to break the epigenetic code. Nat Rev Clin Oncol. 2025;22(2):117–33. doi:10.1038/s41571-024-00972-1. [Google Scholar] [PubMed] [CrossRef]
287. Zhang Z, Zhang Q, Xie J, Zhong Z, Deng C. Enzyme-responsive micellar JQ1 induces enhanced BET protein inhibition and immunotherapy of malignant tumors. Biomater Sci. 2021;9(20):6915–26. doi:10.1039/d1bm00724f. [Google Scholar] [PubMed] [CrossRef]
288. Li L, Meng Y, Wu X, Li J, Sun Y. Bromodomain-containing protein 4 inhibitor JQ1 promotes melanoma cell apoptosis by regulating mitochondrial dynamics. Cancer Sci. 2021;112(10):4013–25. doi:10.1111/cas.15061. [Google Scholar] [PubMed] [CrossRef]
289. Lorusso C, De Summa S, Pinto R, Danza K, Tommasi S. miRNAs as Key players in the management of cutaneous melanoma. Cells. 2020;9(2):415. doi:10.3390/cells9020415. [Google Scholar] [PubMed] [CrossRef]
290. Embaby A, Huijberts SCFA, Wang L, Leite de Oliveira R, Rosing H, Nuijen B, et al. A proof-of-concept study of sequential treatment with the HDAC inhibitor vorinostat following BRAF and MEK inhibitors in BRAFV600-mutated melanoma. Clin Cancer Res. 2024;30(15):3157–66. doi:10.1158/1078-0432.CCR-23-3171. [Google Scholar] [PubMed] [CrossRef]
291. Tao H, Jin C, Zhou L, Deng Z, Li X, Dang W, et al. PRMT1 inhibition activates the interferon pathway to potentiate antitumor immunity and enhance checkpoint blockade efficacy in melanoma. Cancer Res. 2024;84(3):419–33. doi:10.1158/0008-5472.CAN-23-1082. [Google Scholar] [PubMed] [CrossRef]
292. Liang H, Ward WF. PGC-1α: a key regulator of energy metabolism. Adv Physiol Educ. 2006;30(4):145–51. doi:10.1152/advan.00052.2006. [Google Scholar] [PubMed] [CrossRef]
293. Liang J, Yu D, Luo C, Bennett C, Jedrychowski M, Gygi SP, et al. Epigenetic suppression of PGC1α (PPARGC1A) causes collateral sensitivity to HMGCR-inhibitors within BRAF-treatment resistant melanomas. Nat Commun. 2023;14(1):3251. doi:10.1038/s41467-023-38968-7. [Google Scholar] [PubMed] [CrossRef]
294. Hellmann MD, Jänne PA, Opyrchal M, Hafez N, Raez LE, Gabrilovich DI, et al. Entinostat plus pembrolizumab in patients with metastatic NSCLC previously treated with anti-PD-(L)1 therapy. Clin Cancer Res. 2021;27(4):1019–28. doi:10.1158/1078-0432.CCR-20-3305. [Google Scholar] [PubMed] [CrossRef]
295. Huijbers EJM, Khan KA, Kerbel RS, Griffioen AW. Tumors resurrect an embryonic vascular program to escape immunity. Sci Immunol. 2022;7(67):eabm6388. doi:10.1126/sciimmunol.abm6388. [Google Scholar] [PubMed] [CrossRef]
296. Nagy JA, Chang SH, Dvorak AM, Dvorak HF. Why are tumour blood vessels abnormal and why is it important to know? Br J Cancer. 2009;100:865–9. doi:10.1038/sj.bjc.6604929. [Google Scholar] [PubMed] [CrossRef]
297. Barsoum IB, Koti M, Siemens DR, Graham CH. Mechanisms of hypoxia-mediated immune escape in cancer. Cancer Res. 2014;74:7185–90. doi:10.1158/0008-5472.CAN-14-2598. [Google Scholar] [PubMed] [CrossRef]
298. Carter CA, Oronsky BT, Roswarski J, Oronsky AL, Oronsky N, Scicinski J, et al. No patient left behind: the promise of immune priming with epigenetic agents. Oncoimmunology. 2017;6(10):e1315486. doi:10.1080/2162402X.2017.1315486. [Google Scholar] [PubMed] [CrossRef]
299. Kareva I. A combination of immune checkpoint inhibition with metronomic chemotherapy as a way of targeting therapy-resistant cancer cells. Int J Mol Sci. 2017;18(10):2134. doi:10.3390/ijms18102134. [Google Scholar] [PubMed] [CrossRef]
300. Woods DM, Sodré AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res. 2015;3(12):1375–85. doi:10.1158/2326-6066.CIR-15-0077-T. [Google Scholar] [PubMed] [CrossRef]
301. Noviello TMR, Di Giacomo AM, Caruso FP, Covre A, Mortarini R, Scala G, et al. Guadecitabine plus ipilimumab in unresectable melanoma: five-year follow-up and integrated multi-omic analysis in the phase 1b NIBIT-M4 trial. Nat Commun. 2023;14(1):5914. doi:10.1038/s41467-023-40994-4. [Google Scholar] [PubMed] [CrossRef]
302. Miñoza JMA, Rico JA, Zamora PRF, Bacolod M, Laubenbacher R, Dumancas GG, et al. Biomarker discovery for meta-classification of melanoma metastatic progression using transfer learning. Genes. 2022;13(12):2303. doi:10.3390/genes13122303. [Google Scholar] [PubMed] [CrossRef]
303. Zhao S, Li Z, Liu K, Wang G, Wang Q, Yu H, et al. Combining multi-omics analysis with machine learning to uncover novel molecular subtypes, prognostic markers, and insights into immunotherapy for melanoma. BMC Cancer. 2025;25(1):630. doi:10.1186/s12885-025-14012-3. [Google Scholar] [PubMed] [CrossRef]
304. Korfiati A, Grafanaki K, Kyriakopoulos GC, Skeparnias I, Georgiou S, Sakellaropoulos G, et al. Revisiting miRNA association with melanoma recurrence and metastasis from a machine learning point of view. Int J Mol Sci. 2022;23(3):1299. doi:10.3390/ijms23031299. [Google Scholar] [PubMed] [CrossRef]
305. Deng Z, Liu J, Yu YV, Jin YN. Machine learning-based identification of an immunotherapy-related signature to enhance outcomes and immunotherapy responses in melanoma. Front Immunol. 2024;15:1451103. doi:10.3389/fimmu.2024.1451103. [Google Scholar] [PubMed] [CrossRef]
306. Wan G, Nguyen N, Liu F, DeSimone MS, Leung BW, Rajeh A, et al. Prediction of early-stage melanoma recurrence using clinical and histopathologic features. NPJ Precis Oncol. 2022;6(1):79. doi:10.1038/s41698-022-00321-4. [Google Scholar] [PubMed] [CrossRef]
307. Rubinstein JC, Domanskyi S, Sheridan TB, Sanderson B, Park S, Kaster J, et al. Spatiotemporal profiling defines persistence and resistance dynamics during targeted treatment of melanoma. Cancer Res. 2025;85(5):987–1002. doi:10.1158/0008-5472.CAN-24-0690. [Google Scholar] [PubMed] [CrossRef]
308. Kinker GS, Greenwald AC, Tal R, Orlova Z, Cuoco MS, McFarland JM, et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet. 2020;52(11):1208–18. doi:10.1038/s41588-020-00726-6. Epub. [Google Scholar] [PubMed] [CrossRef]
309. Raos D, Ulamec M, Katusic Bojanac A, Bulic-Jakus F, Jezek D, Sincic N. Epigenetically inactivated RASSF1A as a tumor biomarker. Bosn J Basic Med Sci. 2021;21(4):386–97. doi:10.17305/bjbms.2020.5219. [Google Scholar] [PubMed] [CrossRef]
310. Karachaliou GS, Alkallas R, Carroll SB, Caressi C, Zakria D, Patel NM, et al. The clinical significance of adenomatous polyposis coli (APC) and catenin Beta 1 (CTNNB1) genetic aberrations in patients with melanoma. BMC Cancer. 2022;22(1):38. doi:10.1186/s12885-021-08908-z. [Google Scholar] [PubMed] [CrossRef]
311. Kaveti A, Sullivan RJ, Tsao H. KIT-Mutant melanoma: understanding the pathway to personalized therapy. Cancers. 2025;17(22):3644. doi:10.3390/cancers17223644. [Google Scholar] [PubMed] [CrossRef]
312. Larkin J, Marais R, Porta N, Gonzalez de Castro D, Parsons L, Messiou C, et al. Nilotinib in KIT-driven advanced melanoma: results from the phase II single-arm NICAM trial. Cell Rep Med. 2024;5(3):101435. doi:10.1016/j.xcrm.2024.101435. [Google Scholar] [PubMed] [CrossRef]
313. Arroyo Villora S, Castellanos Silva P, Zenz T, Kwon JS, Schlaudraff N, Nitaj D, et al. Biomarker RIPK3 is silenced by hypermethylation in melanoma and epigenetic editing reestablishes its tumor suppressor function. Genes. 2024;15(2):175. doi:10.3390/genes15020175. [Google Scholar] [CrossRef]
314. Morelli M, Madonna S, Albanesi C. SOCS1 and SOCS3 as key checkpoint molecules in the immune responses associated to skin inflammation and malignant transformation. Front Immunol. 2024;15:1393799. doi:10.3389/fimmu.2024.1393799. [Google Scholar] [PubMed] [CrossRef]
315. Nada HR, Rashed LA, Salman OO, Abdallah NMA, Abdelhady MM. Tissue levels of suppressor of cytokine signaling-3 (SOCS-3) in mycosis fungoides. Arch Dermatol Res. 2023;315(2):165–71. doi:10.1007/s00403-022-02339-x. [Google Scholar] [PubMed] [CrossRef]
316. Sutton SK, Koach J, Tan O, Liu B, Carter DR, Wilmott J, et al. TRIM16 inhibits proliferation and migration through regulation of interferon beta 1 in melanoma cells. Oncotarget. 2014;5:10127–39. doi:10.18632/oncotarget.2466. [Google Scholar] [PubMed] [CrossRef]
317. Inamdar GS, Madhunapantula SV, Robertson GP. Targeting the MAPK pathway in melanoma: why some approaches succeed and other fail. Biochem Pharmacol. 2010;80(5):624–37. doi:10.1016/j.bcp.2010.04.029. [Google Scholar] [PubMed] [CrossRef]
318. Gao L, Smit MA, Van den Oord JJ, Goeman JJ, Verdegaal EM, Van der Burg SH, et al. Genome-wide promoter methylation analysis identifies epigenetic silencing of MAPK13 in primary cutaneous melanoma. Pigment Cell Melanoma Res. 2013;26:542–54. doi:10.1111/pcmr.12096. [Google Scholar] [PubMed] [CrossRef]
319. Wang CH, Wang LK, Tsai FM. Exploring potential therapeutic applications of tazarotene: gene regulation mechanisms and effects on melanoma cell growth. Curr Issues Mol Biol. 2025;47(4):237. doi:10.3390/cimb47040237. [Google Scholar] [PubMed] [CrossRef]
320. Abildgaard C, Dahl C, Abdul-Al A, Christensen A, Guldberg P. Inhibition of retinoic acid receptor β signaling confers glycolytic dependence and sensitization to dichloroacetate in melanoma cells. Oncotarget. 2017;8(48):84210–23. doi:10.18632/oncotarget.20476. [Google Scholar] [PubMed] [CrossRef]
321. Hwang S, Kim HE, Min M, Raghunathan R, Panova IP, Munshi R, et al. Epigenetic silencing of SPINT2 promotes cancer cell motility via HGF-MET pathway activation in melanoma. J Invest Dermatol. 2015;135(9):2283–91. doi:10.1038/jid.2015.160. [Google Scholar] [PubMed] [CrossRef]
322. Muthusamy V, Premi S, Soper C, Platt J, Bosenberg M. The hematopoietic stem cell regulatory gene latexin has tumor-suppressive properties in malignant melanoma. J Invest Dermatol. 2013;133(7):1827–33. doi:10.1038/jid.2013.48. [Google Scholar] [PubMed] [CrossRef]
323. Busek P, Duke-Cohan JS, Sedo A. Does DPP-IV inhibition offer new avenues for therapeutic intervention in malignant disease? Cancers. 2022;14(9):2072. doi:10.3390/cancers14092072. [Google Scholar] [PubMed] [CrossRef]
324. Li J, Yang X, Yin C, Li S, Xu Y, Liu B. CDKN2A, a key gene in copper-induced cell death model, influencing melanoma invasion and apoptosis. Discov Oncol. 2025;16(1):246. doi:10.1007/s12672-025-01992-8. [Google Scholar] [PubMed] [CrossRef]
325. Gallagher WM, Bergin OE, Rafferty M, Kelly ZD, Nolan I-M, Fox EJ, et al. Multiple markers for melanoma progression regulated by DNA methylation: insights from transcriptomic studies. Carcinogenesis. 2005;26:1856–67. doi:10.1093/carcin/bgi152. [Google Scholar] [PubMed] [CrossRef]
326. Güvenç C, Neckebroeck F, Antoranz A, Garmyn M, Van den Oord J, Bosisio FM. Bona fide tumor suppressor genes hypermethylated in melanoma: a narrative review. Int J Mol Sci. 2021;22(19):10674. doi:10.3390/ijms221910674. [Google Scholar] [PubMed] [CrossRef]
327. Dika E, Patrizi A, Lambertini M, Manuelpillai N, Fiorentino M, Altimari A, et al. Estrogen receptors and melanoma: a review. Cells. 2019;8(11):1463. doi:10.3390/cells8111463. [Google Scholar] [PubMed] [CrossRef]
328. Caerts D, Garmyn M, Güvenç C. A narrative review of the role of estrogen (Receptors) in melanoma. Int J Mol Sci. 2024;25(11):6251. doi:10.3390/ijms25116251. [Google Scholar] [PubMed] [CrossRef]
329. Körholz J, Chen LS, Strauss T, Schuetz C, Dalpke AH. One gene to rule them all—clinical perspectives of a potent suppressor of cytokine signaling—SOCS1. Front Immunol. 2024;15:1385190. doi:10.3389/fimmu.2024.1385190. [Google Scholar] [PubMed] [CrossRef]
330. Liu S, Ren S, Howell P, Fodstad O, Riker AI. Identification of novel epigenetically modified genes in human melanoma via promoter methylation gene profiling. Pigment Cell Melanoma Res. 2008;21:545–58. doi:10.1111/j.1755-148X.2008.00484.x. [Google Scholar] [PubMed] [CrossRef]
331. Liu S, Howell P, Ren S, Fodstad O, Zhang G, Samant R, et al. Expression and functional analysis of the WAP four disulfide core domain 1 gene in human melanoma. Clin Exp Metastasis. 2009;26:739–49. doi:10.1007/s10585-009-9273-8. [Google Scholar] [PubMed] [CrossRef]
332. Wang N, Zhang H, Cui X, Ma C, Wang L, Liu W. Runx3 induces a cell shape change and suppresses migration and metastasis of melanoma cells by altering a transcriptional profile. Int J Mol Sci. 2021;22(4):2219. doi:10.3390/ijms22042219. [Google Scholar] [PubMed] [CrossRef]
333. Zu T, Wang D, Xu S, Lee CAA, Zhen E, Yoon CH, et al. ATF-3 expression inhibits melanoma growth by downregulating ERK and AKT pathways. Lab Invest. 2021;101(5):636–47. doi:10.1038/s41374-020-00516-y. [Google Scholar] [PubMed] [CrossRef]
334. Wang J, Hua W, Huang SK, Fan K, Takeshima L, Mao Y, et al. RASSF8 regulates progression of cutaneous melanoma through nuclear factor-κb. Oncotarget. 2015;6(30):30165–77. doi:10.18632/oncotarget.5030. [Google Scholar] [PubMed] [CrossRef]
335. Ellmann L, Joshi MB, Resink TJ, Bosserhoff AK, Kuphal S. BRN2 is a transcriptional repressor of CDH13 (T-cadherin) in melanoma cells. Lab Invest. 2012;92(12):1788–800. doi:10.1038/labinvest.2012.140. [Google Scholar] [PubMed] [CrossRef]
336. Cazzato G, Sgarro N, Casatta N, Lupo C, Ingravallo G, Ribatti D. Epigenetics and control of tumor angiogenesis in melanoma: an update with therapeutic implications. Cancers. 2024;16(16):2843. doi:10.3390/cancers16162843. [Google Scholar] [PubMed] [CrossRef]
337. Roider E, Lakatos AIT, McConnell AM, Wang P, Mueller A, Kawakami A, et al. MITF regulates IDH1, NNT, and a transcriptional program protecting melanoma from reactive oxygen species. Sci Rep. 2024;14(1):21527. doi:10.1038/s41598-024-72031-9. [Google Scholar] [PubMed] [CrossRef]
338. Hoek KS, Eichhoff OM, Schlegel NC, Döbbeling U, Kobert N, Schaerer L, et al. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008;68(3):650–6. doi:10.1158/0008-5472.CAN-07-2491. [Google Scholar] [PubMed] [CrossRef]
339. Umar SA, Dong B, Nihal M, Chang H. Frizzled receptors in melanomagenesis: from molecular interactions to target identification. Front Oncol. 2022;12:1096134. doi:10.3389/fonc.2022.1096134. [Google Scholar] [PubMed] [CrossRef]
340. Behrmann I, Wallner S, Komyod W, Heinrich PC, Schuierer M, Buettner R, et al. Characterization of methylthioadenosin phosphorylase (MTAP) expression in malignant melanoma. Am J Pathol. 2003;163:683–90. doi:10.1016/S0002-9440(10)63695-4. [Google Scholar] [PubMed] [CrossRef]
341. Venza M, Visalli M, Catalano T, Biondo C, Beninati C, Teti D, et al. DNA methylation-induced E-cadherin silencing is correlated with the clinicopathological features of melanoma. Oncol Rep. 2016;35:2451–60. doi:10.3892/or.2016.4618. [Google Scholar] [PubMed] [CrossRef]
342. Rubtsova SN, Zhitnyak IY, Gloushankova NA. Dual role of E-cadherin in cancer cells. Tissue Barriers. 2022;10(4):2005420. doi:10.1080/21688370.2021.2005420. [Google Scholar] [PubMed] [CrossRef]
343. Staebler S, Hoechst S, Thongmao A, Schneider N, Bosserhoff AK, Kuphal S. The role of T-Cadherin (CDH13) in treatment options with garcinol in melanoma. Cancers. 2024;16(10):1853. doi:10.3390/cancers16101853. [Google Scholar] [PubMed] [CrossRef]
344. Martinoli C, Gandini S, Luise C, Mazzarol G, Confalonieri S, Giuseppe Pelicci P, et al. Maspin expression and melanoma progression: a matter of sub-cellular localization. Mod Pathol. 2014;27(3):412–9. doi:10.1038/modpathol.2013.157. [Google Scholar] [PubMed] [CrossRef]
345. Chen H, Zheng Z, Kim KY, Jin X, Roh MR, Jin Z. Hypermethylation and downregulation of glutathione peroxidase 3 are related to pathogenesis of melanoma. Oncol Rep. 2016;36:2737–44. doi:10.3892/or.2016.5071. [Google Scholar] [PubMed] [CrossRef]
346. Lindner DJ, Wu Y, Haney R, Jacobs BS, Fruehauf JP, Tuthill R, et al. Thrombospondin-1 expression in melanoma is blocked by methylation and targeted reversal by 5-Aza-deoxycytidine suppresses angiogenesis. Matrix Biol. 2013;32(2):123–32. doi:10.1016/j.matbio.2012.11.010. [Google Scholar] [PubMed] [CrossRef]
347. Carmona FJ, Villanueva A, Vidal A, Muñoz C, Puertas S, Penin RM, et al. Epigenetic disruption of cadherin-11 in human cancer metastasis. J Pathol. 2012;228(2):230–40. doi:10.1002/path.4011. [Google Scholar] [PubMed] [CrossRef]
348. Kilmister EJ, Tan ST. Cancer stem cells and the renin-angiotensin system in the tumor microenvironment of melanoma: implications on current therapies. Int J Mol Sci. 2025;26(3):1389. doi:10.3390/ijms26031389. [Google Scholar] [PubMed] [CrossRef]
349. Vázquez-Naharro A, Bustos-Tauler J, Floristán A, Yuste L, Oltra SS, Vinyals A, et al. Loxl3 promotes melanoma progression and dissemination influencing cell plasticity and survival. Cancers. 2022;14(5):1200. doi:10.3390/cancers14051200. [Google Scholar] [PubMed] [CrossRef]
350. De Araujo ES, Kashiwabara AY, Achatz MI, Moredo LF, De Sa BC, Duprat JP, et al. LINE-1 hypermethylation in peripheral blood of cutaneous melanoma patients is associated with metastasis. Melanoma Res. 2015;25:173–7. doi:10.1097/cmr.0000000000000141. [Google Scholar] [PubMed] [CrossRef]
351. Bozdogan O, Guresci S, Öcalan D, Bozdogan N. Kindlin-3 and RASSF6 are probable biomarkers for predicting metastasis in cutaneous melanoma. Pol J Pathol. 2021;72(3):237–44. doi:10.5114/pjp.2021.111774. [Google Scholar] [PubMed] [CrossRef]
352. Shin Y, Kim S, Ghate NB, Rhie SK, An W. MMP-9 drives the melanomagenic transcription program through histone H3 tail proteolysis. Oncogene. 2022;41(4):560–70. doi:10.1038/s41388-021-02109-5. [Google Scholar] [PubMed] [CrossRef]
353. Salemi R, Falzone L, Madonna G, Polesel J, Cinà D, Mallardo D, et al. MMP-9 as a candidate marker of response to BRAF inhibitors in melanoma patients with BRAFV600E mutation detected in circulating-free DNA. Front Pharmacol. 2018;9:856. doi:10.3389/fphar.2018.00856. [Google Scholar] [PubMed] [CrossRef]
354. Gao L, Van den Hurk K, Nsengimana J, Laye JP, Van den Oord JJ, Beck S, et al. Prognostic significance of promoter hypermethylation and diminished gene expression of SYNPO2 in melanoma. J Invest Dermatol. 2015;135(9):2328–31. doi:10.1038/jid.2015.163. [Google Scholar] [PubMed] [CrossRef]
355. Ngo TKN, Wu HL, Kuo CH, Tu TY. Studying the role of thrombomodulin-plasminogen interaction in spatial and interfacial invasion of melanoma metastatic progression. Int J Biol Macromol. 2025;284(Pt 1):138053. doi:10.1016/j.ijbiomac.2024.138053. [Google Scholar] [PubMed] [CrossRef]
356. Sutton SK, Cheung BB, Massudi H, Tan O, Koach J, Mayoh C, et al. Heterozygous loss of keratinocyte TRIM16 expression increases melanocytic cell lesions and lymph node metastasis. J Cancer Res Clin Oncol. 2019;145:2241–50. doi:10.1007/s00432-019-02981-5. [Google Scholar] [PubMed] [CrossRef]
357. Arab K, Smith LT, Gast A, Weichenhan D, Huang JP, Claus R, et al. Epigenetic deregulation of TCF21 inhibits metastasis suppressor KISS1 in metastatic melanoma. Carcinogenesis. 2011;32(10):1467–73. doi:10.1093/carcin/bgr138. [Google Scholar] [PubMed] [CrossRef]
358. Yamamoto Y, Matsusaka K, Fukuyo M, Rahmutulla B, Matsue H, Kaneda A. Higher methylation subtype of malignant melanoma and its correlation with thicker progression and worse prognosis. Cancer Med. 2020;9(19):7194–204. doi:10.1002/cam4.3127. [Google Scholar] [PubMed] [CrossRef]
359. Walesch SK, Richter AM, Helmbold P, Dammann RH. Claudin11 Promoter hypermethylation is frequent in malignant melanoma of the skin, but uncommon in nevus cell nevi. Cancers. 2015;7(3):1233–43. doi:10.3390/cancers7030834. [Google Scholar] [PubMed] [CrossRef]
360. Huang FJ, Steeg PS, Price JE, Chiu WT, Chou PC, Xie K, et al. Molecular basis for the critical role of suppressor of cytokine signaling-1 in melanoma brain metastasis. Cancer Res. 2008;68(23):9634–42. doi:10.1158/0008-5472.can-08-1429. [Google Scholar] [PubMed] [CrossRef]
361. Helmbold P, Richter AM, Walesch S, Skorokhod A, Marsch WC, Enk A, et al. RASSF10 promoter hypermethylation is frequent in malignant melanoma of the skin but uncommon in nevus cell nevi. J Invest Dermatol. 2012;132(3 Part 1):687–94. doi:10.1038/jid.2011.380. [Google Scholar] [PubMed] [CrossRef]
362. Monson KR, Ferguson R, Handzlik JE, Xiong J, Dagayev S, Morales L, et al. Tyrosine protein kinase SYK-related gene signature in baseline immune cells associated with adjuvant immunotherapy-induced immune-related adverse events in melanoma. Clin Cancer Res. 2024;30(19):4412–23. doi:10.1158/1078-0432.CCR-24-0900. [Google Scholar] [PubMed] [CrossRef]
363. Amaro A, Reggiani F, Fenoglio D, Gangemi R, Tosi A, Parodi A, et al. Guadecitabine increases response to combined anti-CTLA-4 and anti-PD-1 treatment in mouse melanoma in vivo by controlling T-cells, myeloid derived suppressor and NK cells. J Exp Clin Cancer Res. 2023;42(1):67. doi:10.1186/s13046-023-02628-x. [Google Scholar] [PubMed] [CrossRef]
364. Xia C, Leon-Ferre R, Laux D, Deutsch J, Smith BJ, Frees M, et al. Treatment of resistant metastatic melanoma using sequential epigenetic therapy (decitabine and panobinostat) combined with chemotherapy (temozolomide). Cancer Chemother Pharmacol. 2014;74(4):691–7. doi:10.1007/s00280-014-2501-1. [Google Scholar] [PubMed] [CrossRef]
365. Haas NB, Quirt I, Hotte S, McWhirter E, Polintan R, Litwin S, et al. Phase II trial of vorinostat in advanced melanoma. Invest New Drugs. 2014;32(3):526–34. doi:10.1007/s10637-014-0066-9. [Google Scholar] [PubMed] [CrossRef]
366. Huijberts S, Wang L, de Oliveira RL, Rosing H, Nuijen B, Beijnen J, et al. Vorinostat in patients with resistant BRAFV600E-mutated advanced melanoma: a proof of concept study. Future Oncol. 2020;16(11):619–29. doi:10.2217/fon-2020-0023. [Google Scholar] [PubMed] [CrossRef]
367. Jespersen H, Olofsson Bagge R, Ullenhag G, Carneiro A, Helgadottir H, Ljuslinder I, et al. Concomitant use of pembrolizumab and entinostat in adult patients with metastatic uveal melanoma (PEMDAC studyprotocol for a multicenter phase II open label study. BMC Cancer. 2019;19(1):415. doi:10.1186/s12885-019-5623-3. [Google Scholar] [PubMed] [CrossRef]
368. Ny L, Jespersen H, Karlsson J, Alsén S, Filges S, All-Eriksson C, et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat Commun. 2021;12(1):5155. doi:10.1038/s41467-021-25332-w. [Google Scholar] [PubMed] [CrossRef]
369. Weber JS, Levinson BA, Laino AS, Pavlick AC, Woods DM. Clinical and immune correlate results from a phase 1b study of the histone deacetylase inhibitor mocetinostat with ipilimumab and nivolumab in unresectable stage III/IV melanoma. Melanoma Res. 2022;32(5):324–33. doi:10.1097/CMR.0000000000000818. [Google Scholar] [PubMed] [CrossRef]
370. Reijers ILM, Rao D, Versluis JM, Menzies AM, Dimitriadis P, Wouters MW, et al. IFN-γ signature enables selection of neoadjuvant treatment in patients with stage III melanoma. J Exp Med. 2023;220(5):e20221952. doi:10.1084/jem.20221952. [Google Scholar] [PubMed] [CrossRef]
371. Hong DS, Kang YK, Borad M, Sachdev J, Ejadi S, Lim HY, et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br J Cancer. 2020;122(11):1630–7. doi:10.1038/s41416-020-0802-1. [Google Scholar] [PubMed] [CrossRef]
372. Beg MS, Brenner AJ, Sachdev J, Borad M, Kang YK, Stoudemire J, et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs. 2017;35(2):180–8. doi:10.1007/s10637-016-0407-y. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools