
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Role of necroptosis in spinal cord injury and its therapeutic implications
1 The Affiliated Hospital 2 of Nantong University, Nantong University, The First People’s Hospital of Nantong, Nantong, 226001, China
2 Key Laboratory for Restoration Mechanism and Clinical Translation of Spinal Cord Injury, Nantong, 226001, China
3 Research Institute for Spine and Spinal Cord Disease of Nantong University, Nantong, 226001, China
* Corresponding Author: ZHIMING CUI. Email:
# Jiawei Fu and Chunshuai Wu should be considered joint first authors
(This article belongs to the Special Issue: Neuroimmune Interactions at the Crossroads of Health and Disease)
BIOCELL 2023, 47(4), 739-749. https://doi.org/10.32604/biocell.2023.026881
Received 30 September 2022; Accepted 26 December 2022; Issue published 08 March 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Spinal cord injury (SCI), a complex neurological disorder, triggers a series of devastating neuropathological events such as ischemia, oxidative stress, inflammatory events, neuronal apoptosis, and motor dysfunction. However, the classical necrosome, which consists of receptor-interacting protein (RIP)1, RIP3, and mixed-lineage kinase domain-like protein, is believed to control a novel type of programmed cell death called necroptosis, through tumour necrosis factor-alpha/tumour necrosis factor receptor-1 signalling or other stimuli. Several studies reported that necroptosis plays an important role in neural cell damage, release of intracellular pro-inflammatory factors, lysosomal dysfunction and endoplasmic reticulum stress. Recent research indicates that necroptosis is crucial to the pathophysiology of a number of neurological disorders and SCIs. In our review, we summarize the potential role of programmed cell death regulated by necroptosis in SCI based on its molecular and pathophysiological mechanisms. We also summarize the targets of several necroptosis pathways, which provide a more reliable reference for the treatment of SCI.Keywords
Spinal cord injury (SCI), a common clinical traumatic condition with high mortality and disability rates, is typically caused by falls and traffic accidents (Ihalainen et al., 2017). Secondary injury is initiated within minutes of the primary SCI, leading to vascular rupture and haemorrhage, local ischemia and destruction of neural tissue in the area of injury, thereby causing a state of spinal shock. The disruption of the microenvironment, release of inflammatory factors, ion imbalance, excessive production of free radicals and release of excitotoxic substances lead to pathological apoptosis and proliferation and activation of neurons, glial cells and oligodendrocytes in the central nervous system of the spinal cord. This is followed by a series of clinical symptoms, such as neurological dysfunction (O’Shea et al., 2017; Bellver-Landete et al., 2019).
Necrosis and apoptosis are currently known to be the predominant modes of cell death in SCI (Beattie et al., 2002; Xu et al., 2018). Previous studies suggested that necrosis is a passive death of cells that is not regulated by genes. However, recent studies showed that necrosis, like apoptosis, is strictly regulated by genes, and therefore, this genetically regulated form of necrosis is termed necroptosis (Degterev et al., 2005). The morphological features of necroptosis are similar to those of necrosis, such as cell swelling, cell membrane rupture, organelle disintegration and protein denaturation. In addition, there are some specific morphological and pathophysiological signs of necroptosis: (1) the presence of autophagy in the necrotic terminals, (2) an increase in the reactive oxygen radicals in some necrosis-like cells and (3) its inhibition by the small molecule, receptor-interacting protein (RIP)-specific inhibitor, Necrostatin-1 (Nec-1) (Han et al., 2007; Mehta et al., 2007; Ying and Padanilam, 2016). Necroptosis can cause an inflammatory response, but its cell death is programmed in a gene-regulated RIP-dependent manner. Necroptosis is not regulated by a caspase-dependent pathway (Hitomi et al., 2008; Dunai et al., 2011). Necroptosis is associated with various diseases, including tumours, ischemic brain injury, neurodegenerative diseases and viral myocardial infarction (Degterev et al., 2005; Smith et al., 2007; Gong et al., 2019).
Generally, as shown in Fig. 1, the necroptotic pathway can be activated by several stimuli, including the tumour necrosis factor receptor (TNFR) superfamily, pattern recognition receptors (PRRs), T cell receptors and various chemotherapeutic agents (Lalaoui et al., 2015). Among them, the TNF-alpha (TNF-α)/TNFR signalling is the most classical and well-studied pathway (Fulda, 2013). Necroptosis is a part of an intracellular signalling pathway transduced by RIP1/3 and mixed lineage kinase domain-like protein (MLKL) (Sun et al., 2012). TNF binding to TNFR1 causes conformational changes in TNFR1 trimers, leading to the recruitment of a variety of proteins, including RIPK1, TNFR-associated death domain, a cellular inhibitor of apoptosis protein and TNFR-associated factor (Shan et al., 2018; Yu et al., 2021; Christgen et al., 2022). RIP1 kinase activity seems essential for necroptosis activation (Holler et al., 2000). RIP1 is an important effector downstream of death receptors and PRRs, controlling the pro-survival, apoptotic, and especially inflammatory necroptotic pathways (Fiani et al., 2021). The binding of TNFR1 to TNF-α leads to the recruitment of RIP1 through its death domain, which activates pro-apoptotic caspase-8. RIP3 and RIP1 interact through the RIP homotypic interaction motif, leading to the formation of necrosomes which activate downstream effector proteins to trigger the necroptosis pathway and inflammatory response described above (Li et al., 2012). MLKL has two structural domains, and the 4HB domain is responsible for the lethal activity of MLKL (Chen et al., 2014b; Hildebrand et al., 2014); the pseudokinase domain (PsKD) structural domain plays an important role in cellular signalling, acting as a dynamic scaffold and helping in protein-protein interaction (Boudeau et al., 2006). Once RIP3 is activated, the PsKD structural domain is phosphorylated and the conformation changes. The 4HB domain is unlocked and MLKL is released from the RIP3 activation platform (Rodriguez et al., 2016; Grootjans et al., 2017).
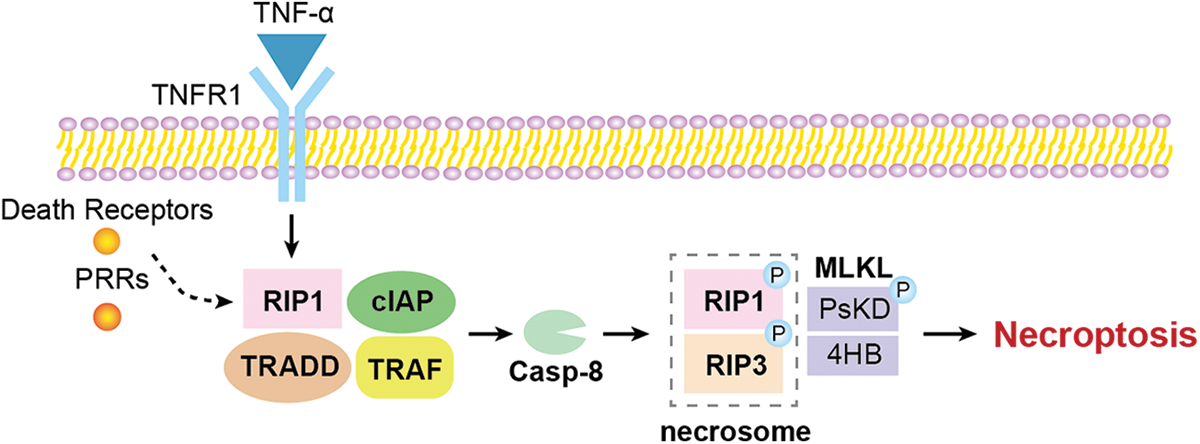
Figure 1: The overview of the molecular mechanism of necroptosis.
Regulatory Role of Necroptosis in Different Neural Cells after Spinal Cord Injury
Current evidence suggests that the necroptosis signalling cascade is critical in neurological diseases and SCI. Kanno et al. (2015) showed that RIP3 protein levels significantly increased at the site of injury after spinal cord hemisection in mice. Twenty-four hours after SCI, they observed an increase in the number of RIP3-positive cells at the site of injury, peaking at 3 days and persisting for at least 3 weeks after injury. As shown in Fig. 2, they also demonstrated for the first time that RIP3 expression is noticeably increased in several neural cells present in the injured spinal cord, including neurons, astrocytes, and oligodendrocytes, suggesting that necroptosis develops in various neural cells and contributes to different pathological mechanisms following SCI. However, in this study, despite the propidium iodide-labelling and terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining of spinal cord sections from the injured mice, the exact pathological mechanisms of necroptosis and the underlying molecular mechanisms are not fully elucidated.
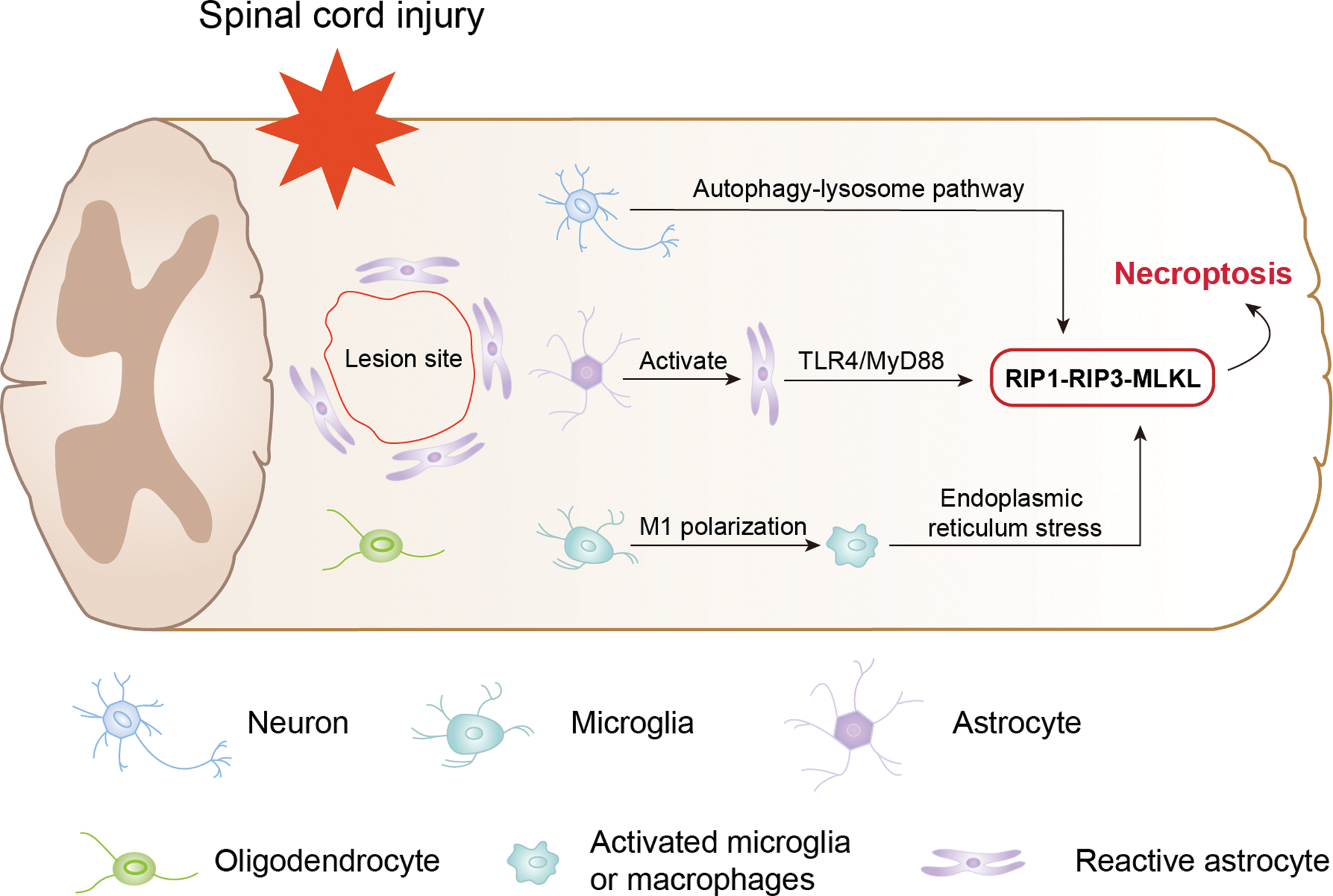
Figure 2: Regulatory role of necroptosis in different neural cells after spinal cord injury.
Wu et al. (2016) found that the proteasome beta-4 subunit (PSMB4) was notably upregulated in neurons after SCI in rats, and double staining showed a significant increase in PSMB4 expression in RIP3-positive neurons, suggesting its important role in RIP3-regulated neuronal necroptosis. Even in vitro experiments showed that overexpression and knockdown of PSMB4 by a plasmid significantly affected the expression of the RIP3/p-MLKL pathway. Liu et al. (2018) reported that necroptosis of neurons after SCI is also regulated by the autophagy-lysosome pathway. They demonstrated that autophagy was suppressed in mice following SCI and the first suppression of autophagic flux was brought on by the quick fall in lysosomal function observed following SCI, as evidenced by a drop in protein levels and lysosomal enzyme activity. They observed an accumulation of necroptotic markers (RIP1, RIP3 and MLKL), particularly in the neurons showing inhibition of autophagic flux and lysosomal damage. In vitro experiments showed that the accumulation of RIP1, RIP3 and MLKL after lysosomal inhibition led to the formation of necrosomes and the onset of necroptosis. The authors treated the injured mice with rapamycin, an inhibitor of the mammalian target of rapamycin (mTOR) and inducer of autophagy and lysosomes, and found that rapamycin reduced the suppression of autophagic flux and RIP1 accumulation. Therefore, in order to prevent necroptosis after SCI, it may be more beneficial to improve or restore lysosomal function.
In addition to the loss of neuronal cells, ongoing tissue loss, reactive astrocyte proliferation and chronic inflammation are common in secondary SCI (Ju et al., 2014; Schwab et al., 2014). Fan et al. (2016) investigated how reactive astrocytes, a major component of glial scarring, function in SCI. First, on day-5 post-injury in mice, they performed double immunostaining with TUNEL, labelled with a yellow fluorescent protein (YFP)-labelled astrocytes in advance. They found that YFP/TUNEL double-positive cells were almost non-existent. Next, they used the markers of autophagy (Lamp2a, light chain 3 [LC3] and Beclin1) to double-label with YFP and found that double-positive cells were still rare. Further, they used PI-labelling with YFP double-staining and found that the number of double-positive cells greatly increased and peaked on day-5 post-injury, suggesting that reactive astrocytes undergo a pathological process of necrosis rather than apoptosis and autophagy. To investigate whether reactive astrocytes undergo necroptosis, the authors double-labelled RIP3 and MLKL with glial fibrillary acidic protein (GFAP) and found that approximately 80% of RIP3-positive cells were GFAP-positive, indicating that after SCI in mice, reactive astrocytes likely experience RIP3/MLKL-mediated necroptosis. Given that M1 microglia/macrophages are a significant source of toxic inflammatory factors following SCI (Kigerl et al., 2009), the authors performed an in vitro model of M1 microglia/macrophages causing astrocyte necroptosis. With conditioned media (CM) collected from microglia and macrophages in various polarization stages, they activated astrocytes. The astrocytes treated with CM from M1 microglia/macrophages showed higher expression of RIP3 and MLKL than the control and M2 groups. Additionally, in astrocytes treated with M1 CM, intracellular ATP was considerably decreased, and the proportion of PI-labelled cells increased. On the contrary, by depleting M1 microglia with gadolinium chloride (GdCl3) at the lesion site (Miron et al., 2013), the authors found that GdCl3 treatment significantly reduced RIP3 and MLKL expression in the spinal cord of mice and resulted in smaller spinal cord cavities and better recovery of motor function. These findings imply that the induction of necroptosis in astrocytes following SCI is significantly influenced by M1 microglia/macrophages. They further explored the necroptotic signalling pathway induced by M1 microglia/macrophages. Previous studies (Kigerl et al., 2007; He et al., 2011; Ransohoff and Brown, 2012; Heiman et al., 2014) have suggested that toll-like receptors (TLRs) have a role in the necroptosis of macrophages and the activation of astrocytes after SCI in vitro, and since the studies showed that most reactive astrocytes were co-labelled with TLR4 by double immunofluorescence (IF), the authors chose to validate the more common TLR4/MyD88 signalling pathway. TLR4 was found to be highly colocalized with RIP3, and MyD88 was also strongly expressed in reactive astrocytes in the injured spinal cord in mice, suggesting that the TLR4/MyD88 signalling pathway is involved in the activation of necroptosis in astrocytes after SCI. In vivo, GdCl3 treatment significantly decreased astrocytes that were expressing TLR4 or MyD88. These experiments suggest that the necroptosis of astrocytes caused by M1 microglia/macrophages in mice is mediated by TLR4/MyD88 signalling. However, this study only verified the role of TLR4 and did not completely exclude the role of other prominent members of the TLR family.
Another study by Fan et al. (2015) investigated the mechanism of microglia-induced necroptosis in the endoplasmic reticulum (ER). They found that MLKL is predominantly expressed in microglia by co-labelling MLKL protein with ionized calcium-binding adapter molecule 1 (Iba-1), GFAP, CC1 or NeuN (markers for microglia, astrocytes, oligodendrocytes and neurons, respectively). Immunohistochemistry and immunoelectron microscopy showed that MLKL was mainly distributed in the cytoplasm and membrane of Iba-1-positive cells. In other words, microglia may have undergone necroptosis after SCI. Immunoelectron microscopy revealed that both MLKL and RIP3 were expressed on ER, suggesting that ER is involved in the pathological process of necroptosis. To investigate whether microglial necroptosis is associated with ER stress, triple immunostaining of MLKL, Iba-1 and GRP78 (ER stress sensor) was performed. Approximately 68.2% of MLKL/Iba-1 double-positive cells exhibited positive GRP78, indicating that ER stress is induced by microglial necroptosis. For in vitro assessment, the authors constructed a cell model of necroptosis by inducing oxygen-glucose deprivation (OGD). OGD treatment induced ER stress as well as necroptosis in microglia; accordingly, 4-phenylbutyric acid (an ER stress inhibitor) effectively blocked GRP78 and p-eIF2a (two ER markers) as well as MLKL and RIP3, suggesting that ER stress is involved in OGD-induced microglial necroptosis.
Inhibiting Necroptosis-Regulated Cell Death for the Therapy of Spinal Cord Injury
In recent years, there is still a shortage of research on programmed cell death (PCD) in SCI, although there is rising evidence that PCD plays a pivotal role after SCI (Chen et al., 2014a; Zha et al., 2014). Scholars have recognized the need to consider therapeutic strategies for SCI to promote spinal cord repair and regeneration while limiting cell death and axonal injury (Festoff, 2014). Thus, inhibition of necroptosis is receiving much attention from researchers and clinicians as a new therapeutic strategy for SCI.
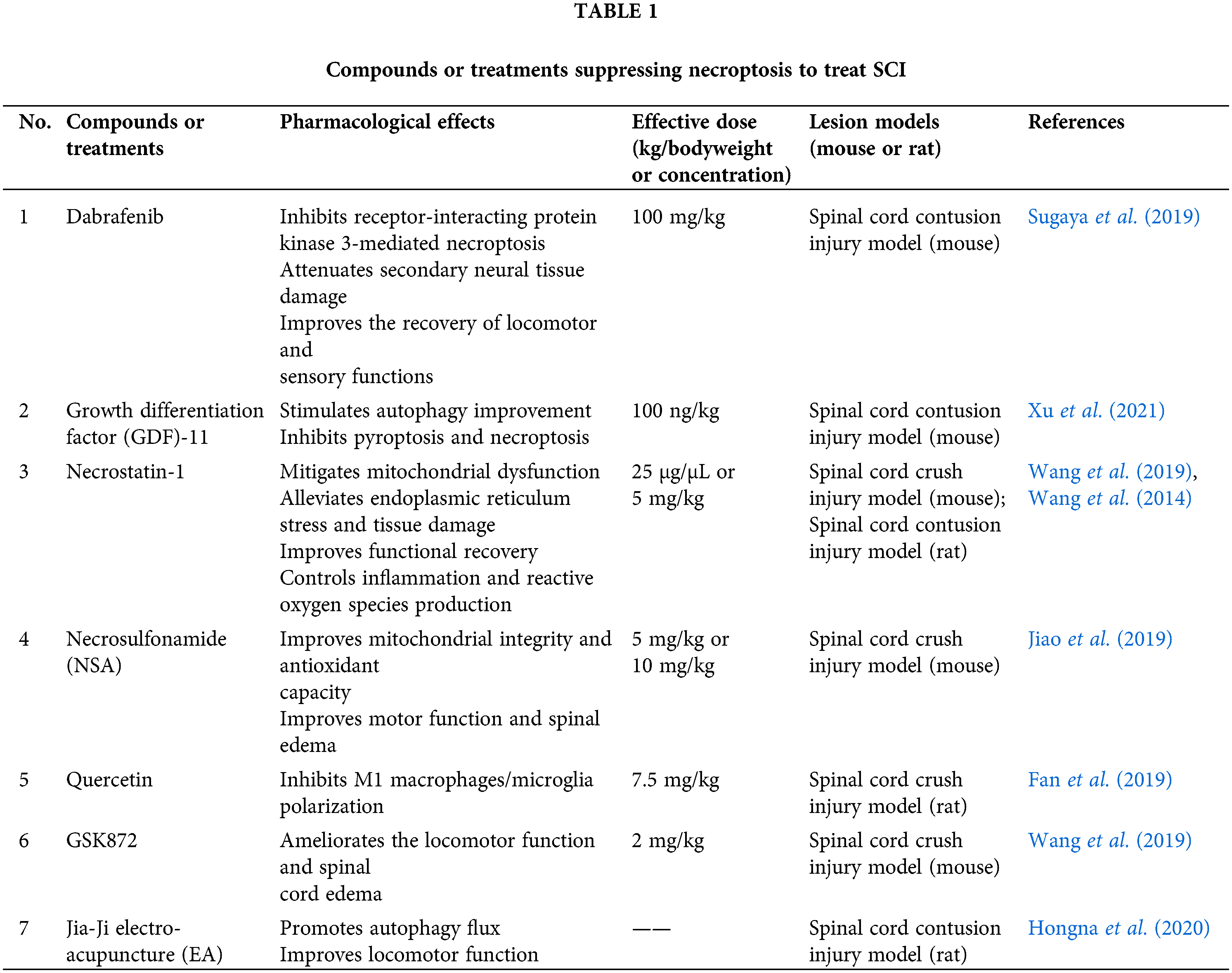
Dabrafenib, a B-RAFV600E inhibitor and an anticancer agent (Al-Jundi et al., 2020; Kam et al., 2021; Lorusso et al., 2021), can selectively inhibit RIP3 (Li et al., 2014). However, the therapeutic effect of dabrafenib on SCI has not been studied yet. Sugaya et al. (2019) treated mice with dabrafenib to assess its effect on the recovery of motor function in mice with the spinal cord contusion injury model, as shown in Fig. 3 and Table 1. The total Basso mouse scale (BMS) score and the BMS sub-score of motor function were consistently higher in the dabrafenib-treated mice than in the control-treated mice after injury. The authors further assessed the motor behaviour of the injured mice using the inclined plane (IP) test. The IP angle was significantly higher in the dabrafenib-treated mice, indicating better recovery from muscle paralysis. Further, the mechanical and thermal allodynia of the hind limbs in mice were evaluated. The von Frey test was used to examine the retraction threshold of the hind paw when the mice were mechanically stimulated, and the Hargreaves test was used to examine the retraction latency of the hind limbs when the mice were thermally stimulated. The dabrafenib-treated mice had longer retraction latency and better thermal sensitivity than the control-treated mice. They also found that the number of p-MLKL positive cells in the spinal cord of dabrafenib-treated mice was significantly lower compared to that in control-treated mice. At the epicentre and 500 µm rostral and caudal from the epicentre, the number of PI-positive cells in the spinal cord sections of mice receiving dabrafenib treatment was considerably less than that of mice undergoing control treatment, indicating that dabrafenib reduces necrotic cell death by inhibiting the activation of the RIP3-MLKL pathway. Dabrafenib also dramatically reduced inflammatory infiltration, vacuolar degeneration and neuronal loss at the site of the lesion in the white matter of the spinal cord of injured mice. Immunostaining of neurofilaments using the anti-RT97 antibody demonstrated that dabrafenib also reduces axonal damage in the damaged spinal cord. Overall, Dabrafenib inhibits RIP3-mediated necroptosis in mice, providing neuroprotection and promoting functional recovery after SCI. However, this study did not establish an in vitro cellular model to further investigate the effects of dabrafenib on neuronal cells.
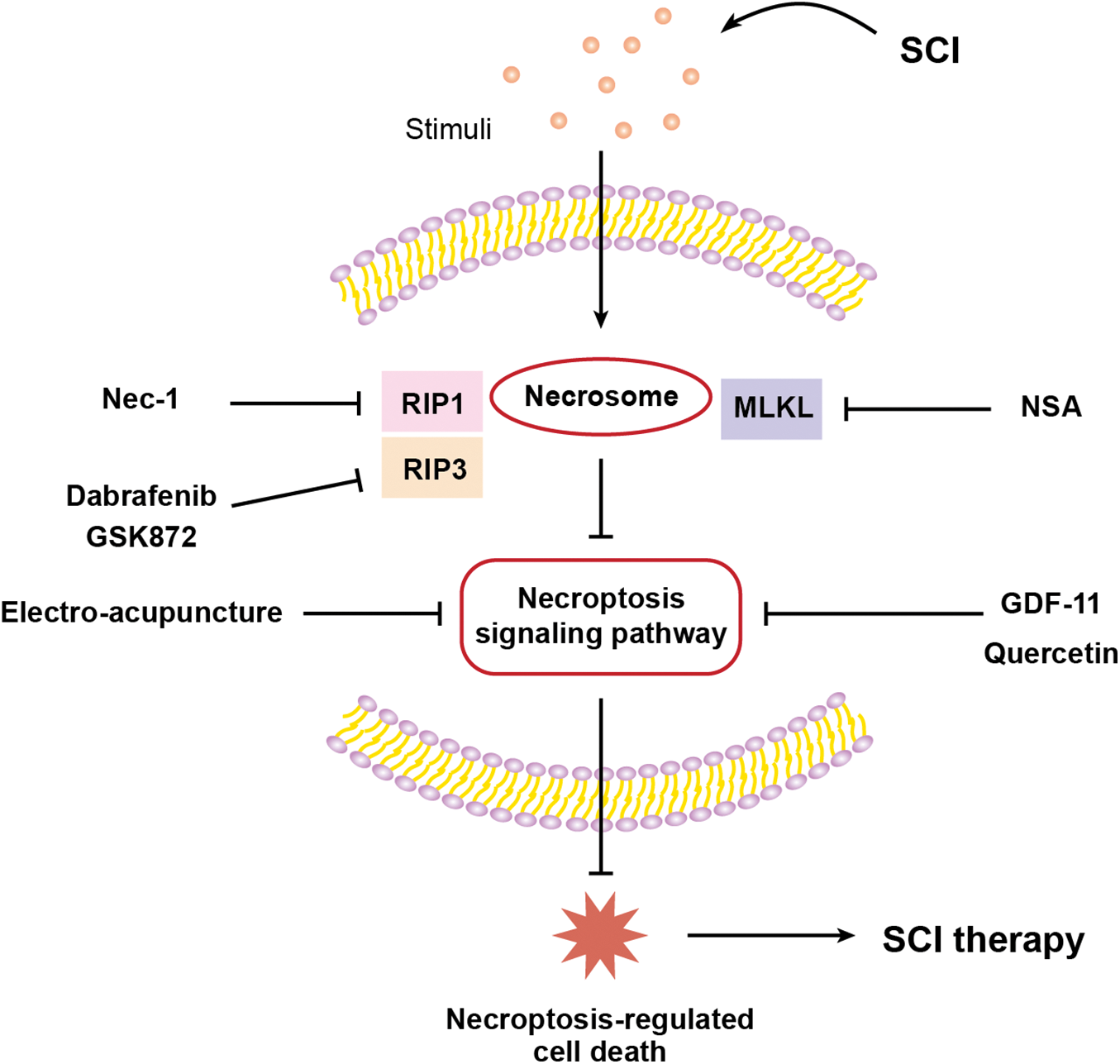
Figure 3: Graphical description of targeting necroptosis-regulated cell death for spinal cord injury therapy.
Growth differentiation factor 11 (GDF-11)
Growth differentiation factor 11, a member of the transforming growth factor-β (TGF-β) superfamily (McPherron et al., 1999; Liu, 2006), is an anti-aging factor that regulates neurogenesis and has neuroprotective effects following cerebral ischemia injury. Many biological processes, including histogenesis, embryonic development, cancer and metabolic disorders, are impacted by its members (Hanna and Frangogiannis, 2019). GDF-11 reduces neuronal apoptosis and protects against cerebral ischemia injury (Zhao et al., 2020). It also ameliorates ischemic stroke in mice by activating autophagy and stimulating neurogenesis and angiogenesis (Hudobenko et al., 2020). To investigate the role of GDF-11 in SCI, Xu et al. (2021) performed western blot analysis and IF to analyse molecular expression related to cell pyroptosis, autophagy and necroptosis. GDF-11 was found to significantly promote the recovery of motor function, increase autophagy, inhibit cell pyroptosis and attenuate necroptosis after SCI with the spinal cord contusion injury model. These neuroprotective effects of GDF-11 can be reversed by the autophagy inhibitor, 3-methyladenine (3MA), suggesting an important role of autophagy in the treatment of SCI. As the expression of transcription factor E3 (TFE3) was significantly higher in the nucleus but lower in the cytoplasm of neurons in the GDF-11-treated group, the authors speculated that the protective effect of GDF-11 is probably related to the activation of TFE3. The expression levels of pyroptosis-associated markers (caspase recruitment domain [ASC], interleukin [IL]-18, IL-1β, gasdermin D [GSDMD], caspase-1, NLR family pyrin domain containing 3 [NLRP3] and NLRP1) and necroptosis-associated markers (RIP1, RIP3 and MLKL) were increased whereas vacuolar protein sorting 34, beclin1, cathepsin D, and Microtubule-associated protein light chain 3 II expression levels were reduced in the TFE3-silenced group; this group also revealed larger glial scar areas, lower microtubule-associated protein 2 expression and fewer synuclein (SYN)-positive synapses. Even BMS motor scores displayed a tendency to decrease in the TFE3-silenced group. These findings demonstrated that GDF-11 exerts its neuroprotective effects by enhancing autophagy and decreasing cell pyroptosis and necroptosis primarily through nuclear translocation and TFE3 activation. The authors also investigated whether the AMP-activated protein kinase (AMPK)-transient receptor potential mucolipin 1 (TRPML1)-calcineurin signalling pathway regulates TFE3 activation following treatment with GDF-11. When the AMPK blocker, compound C (CC), was administered to the GDF-11-treated group, protein expression of p-AMPK and TFE3 nuclear translocation status were found to be significantly lower than those in the control group. Moreover, TRPML1 and calcineurin expression levels were significantly suppressed after the administration of CC. Meanwhile, caspase-1, GSDMD, RIP1, RIP3, and p62 had significantly higher levels of expression in the GDF-11+CC group than in the GDF-11 group, but LC3II had the opposite trend. These critical findings finally illustrated that GDF-11 activates TFE3, enhances autophagy, inhibits cell pyroptosis and necroptosis and ultimately exerts neuroprotective effects through the AMPK-TRPML1-calcineurin signalling cascade. Sutherland et al. (2020), on the other hand, discovered for the first time that GDF-11 is neurotoxic to primary neurons in the acute phase of simulated stroke, mostly via ALK4 receptor signalling. Therefore, when applying GDF-11 treatment, sufficient attention needs to be paid to its neurotoxicity.
Nec-1, a small molecule inhibitor of necroptosis, targets RIP1 (Degterev et al., 2008) and plays an essential role in inhibiting pathological death of various cells and tissues in the central nervous system, including ischemic brain injury and neurodegenerative diseases (Xu et al., 2010; Chavez-Valdez et al., 2012). Studies related to SCI showed that Nec-1 can alleviate necroptosis by reducing mitochondrial dysfunction, mitigating endoplasmic reticulum stress (ERS), effectively controlling inflammation and reactive oxygen species (ROS) production, and eventually exerting neuroprotective functions. Wang et al. (2015) found that Ca2+ concentration in mitochondria significantly increased in the SCI group, whereas mitochondrial membrane potential (MMP) decreased at both 12 and 24 h after SCI, indicating severe mitochondrial membrane damage. After treatment with Nec-1, Ca2+ concentration gradually dropped and MMP remarkably hyperpolarized and incrementally increased, suggesting partial recovery of mitochondrial membranes. Furthermore, regarding the significant downregulation of mitochondrial respiratory chain complex I activity after SCI (22.36 ± 1.55 nmol/L), Nec-1 treatment blocked the release of cytochrome c and resulted in a pronounced increase in mitochondrial respiratory chain complex I activity (26.09 ± 1.87 nmol/L). Moreover, electron microscopy revealed mitochondrial swelling, disruption of the inner and outer mitochondrial membranes, and impairment of the cristae of mitochondria in the SCI group. However, Nec-1 effectively relieved the swelling and restored the mitochondrial structure to normal. Simultaneously, the authors investigated the gene expression of three major transcription factors (PGC-1, NRF-1 and Tfam) necessary for mitochondrial biogenesis and found that Nec-1 reverses the decrease in Tfam after SCI. The effect of Nec-1 on mitochondrial fusion and division was further investigated. Expression of mitochondrial fusion-related genes (Mfn1 and Mfn2) was increased at 6 h after SCI, whereas their expression was remarkably suppressed after Nec-1 treatment; Fis1 gene expression was downregulated after SCI but enhanced after Nec-1 treatment. Altogether, Nec-1 can treat SCI by attenuating mitochondrial dysfunction. In another study by the same authors (Wang et al., 2014), Nec-1 treatment for SCI not only inhibited necroptosis by inhibiting the recruitment of RIP1/3-MLKL but also reduced lesions, inflammatory factors and ROS production in the area of SCI, thereby improving the pathological condition and blood supply.
Wang et al. (2017) demonstrated an increase in the expression of ERS-related genes at 6 and 12 h after SCI, including C/EBP homologous protein (CHOP), immunoglobulin-binding protein (BiP/GRP78) and X box-binding protein-1 (XBP-1); however, these changes were reversed by Nec-1. IF detected that GRP78, CHOP and XBP1 proteins were mainly expressed in the cytoplasm of neurons and astrocytes and less in microglia. Electron microscopy revealed a decrease in the ribosome shedding of neuronal ER and the ER structure was more intact compared to the SCI group after Nec-1 treatment. Therefore, these investigations suggest that Nec-1 can be a promising inhibitor of necroptosis, and it deserves further studies to explore the potential role in the intervention of SCI.
NSA can specifically block MLKL proteins (Sun et al., 2012). NSA has now been applied to treat Alzheimer’s disease (AD), psoriatic inflammation, and acute myeloid leukemia (AML) and protects intervertebral disc degeneration (Duan et al., 2020; Motawi et al., 2020; Zhang et al., 2020; Chen et al., 2022). Wang et al. (2018) demonstrated for the first time that NSA significantly reduces the expression of MLKL in rats with SCI, whereas it did not significantly alter other necroptosis-related factors such as RIP1 and RIP3. Similarly, it did not affect the caspase-3 activity associated with apoptosis. These critical findings suggest that NSA specifically inhibits the activation of MLKL, which ultimately promotes the recovery of motor function in rats with the spinal cord crush injury model. Based on this study, Jiao et al. (2019) investigated the molecular mechanism and therapeutic window behind the inhibition of MLKL by NSA with the same crush model. They showed that OGD-treated neuronal cells exhibit higher cell viability at NSA concentrations of 3 and 10 μM and the increase in cell viability is time-dependent. They further evaluated the role of NSA in mitochondrial capacity and antioxidant capacity in vitro. They demonstrated that NSA restores ATP and MMP levels and significantly inhibits Bcl-2-associated X-protein (Bax) protein expression but increases B-cell lymphoma 2 (Bcl-2) protein expression. Also, NSA decreases the expression of ROS and malonyldialdehyde but increases the expression of superoxide dismutase and glutathione. The former two are commonly used to detect oxidative function, whereas the latter two are used to detect antioxidative function.
To investigate the therapeutic window of NSA in improving motor function, the authors divided the NSA treatment groups into NSA-L (1 mg/kg), NSA-M (5 mg/kg) and NSA-H (10 mg/kg) at different concentrations. The NSA-M and NSA-H groups were found to have significant therapeutic effects from day 7 after SCI, as evidenced by a significant increase in the forelimb grip strength test and BMS score, as well as a pronounced decline in spinal cord water content. Both NSA-M and NSA-H groups exhibited the same neuroprotective function after SCI. In addition, the experiment was divided into 15 min, 30 min, 1, 3, 6, 12 and 24 h postoperative groups according to the time of NSA administration (5 mg/kg). The recovery of forelimb grip strength was found to be optimum for 15 and 30 min postoperative administration on day 7 after SCI. The later the NSA was administered, the slower the recovery of forelimb grip strength. There was also a significant improvement in BMS scores and a decrease in spinal oedema in the 1, 3, 6 and 12 h groups from 21 to 28 days after SCI. These results suggest that in the SCI crush injury model, NSA administration within 12 h after SCI at a concentration of 5 mg/kg (or 10 mg/kg) may be effective in alleviating SCI and improving the mobility of injured mice.
Quercetin, an important flavonoid component of several herbs (Boots et al., 2008; Hogan et al., 2010; Abarikwu et al., 2012), has long been used as an antioxidant and anti-inflammatory agent in herbal medicine. It also reduces neuronal death and inhibits pyroptosis and inflammatory responses in SCI (Jiang et al., 2016). Fan et al. (2019) investigated the effect of quercetin on the survival and macrophages/microglia polarization of oligodendrocytes (OLs) after SCI. They performed double staining of RIP3/CC1, MLKL/CC1 and p-MLKL/CC1 (CC1 is a marker for OLs) 10 days after SCI and found a significant decrease in the number of RIP3/CC1, MLKL/CC1 and p-MLKL/CC1 double positives after quercetin treatment, but a significant increase in CC1 positive cells. It suggests that treatment with quercetin after SCI significantly reduces necroptosis and increases the survival rate of OLs. To determine the effect of quercetin on M1 polarization of macrophages/microglia, Fan et al. (2019) measured the mRNA levels of TNF-α, inducible nitric oxide synthase (iNOS) and CD86 (markers of M1 macrophages/microglia) 10 days after injury and noticed that the mRNA levels of TNF-α, iNOS and CD86 reduced in the quercetin-treated group. IF showed that the number of macrophages/microglia which expressed iNOS in spinal cord sections decreased in the quercetin-treated group. In contrast, quercetin increased the mRNA levels of arginase1, IL-4 and CD206 (markers of M2 polarization). The number of arginase1-positive macrophages/microglia was increased. These notable findings suggest that quercetin inhibits macrophages/microglia polarization of the M1 phenotype and promotes M2 polarization. They cultured OLs and microglia in vitro and induced necroptosis to further investigate the molecular mechanisms. After treatment with M1 microglia-CM, ROS levels in OLs and PI-labelled cells increased, while ATP levels decreased, indicating the occurrence of necroptosis. Moreover, M1 CM significantly enhanced the expression of RIP3, MLKL and p-MLKL, which was blocked by quercetin-modified M1 CM, whereas quercetin alone had no remarkable effect on necroptosis in OLs. Detection of mRNA levels of M1- and M2-related markers revealed that quercetin decreased the mRNA levels of TNF-α, IL-12 and IL-1β but increased the mRNA levels of IL-4, IL-10 and TGF-β in M1 microglia. These significant findings suggest that quercetin inhibits M1 polarization in macrophages/microglia, which in turn inhibits necroptosis in OLs. Furthermore, signal transducer and activator of transcription 1 (STAT1) and nuclear factor (NF)-κB signalling pathways were found to be involved in the biological process of quercetin-regulated necroptosis in OLs. Altogether, in vivo and in vitro, quercetin markedly reverses the high expression of iNOS, p-STAT1, NF-κB and p-NF-κB caused by SCI.
GSK872, a specific inhibitor of RIP3, can significantly inhibit RIP3 activity and its downstream events (Chen et al., 2018). Wang et al. (2019) reported that SCI can lead to mitochondrial dysfunction in mice, such as a reduction in ATP as well as a decrease in antioxidant capacity and MMP. However, GSK872 improved the levels of ATP and MMP and increased the antioxidant capacity. In vitro, OGD-induced neuronal cells were treated with GSK872 to investigate the cytoprotective effects of GSK872. Experiments using Cell Counting Kit-8 showed that GSK872 (3 and 10 μM) effectively improves the cell viability of OGD-induced spinal cord neurons. Dual IF showed that GSK872 efficiently reduces the expression of p-RIP3 in neuronal cells. Moreover, GSK872 markedly increased the levels of ATP and MMP in neuronal cells. The upregulation of Bcl-2 and decrease in the protein expression of Bax indicated the protective role of GSK872 in OGD-induced mitochondrial dysfunction in spinal cord neurons.
EA, a traditional method in Chinese medicine wherein needles are inserted into acupuncture points and connected to a micro-pulse current to produce synthetic EA stimulation, is proven to be beneficial for neurological and functional recovery after SCI (Ding et al., 2011; Huang et al., 2011). However, its working mechanism is not fully elucidated. Hongna et al. (2020) reported that treatment with Jia-Ji EA, 6 h after SCI with the spinal cord contusion injury model, significantly enhances the Basso-Beattie-Bresnahan (BBB) scores, reduces spinal cord hematoma and necrotic cavities, ameliorates neuronal swelling and decreases neuronal loss and inflammatory cell infiltration in rats. They also showed that EA decreases the expression of necroptosis-related markers (RIP1, RIP3 and MLKL), promotes LC3 expression, reduces P62 expression, increases autophagosome formation and accelerates autophagic flux. When chloroquine (CQ), a lysosomal inhibitor, was added to EA treatment, it remarkably decreased the BBB motor scores at 3 and 7 days after SCI with a significant reduction in the number of neurons. Electron microscopy revealed a much greater increase in autophagosomes and a decrease in autolysosomes in EA + CQ group than in the EA group. The effect of EA on RIP1, RIP3, and MLKL protein expression was partially reversed by CQ, which facilitated the development of necrotic complexes and increased neuronal necrosis.
Conclusion and Future Directions
A complex series of molecular cascade reactions occur after SCI, causing damage to various neural cells, thus limiting the effectiveness of therapeutic interventions in the injured region. In this review, we summarized the mechanisms of necroptosis as one of the programmed cell deaths in SCI and the regulatory role of necroptosis in different neural cells (e.g., neurons, microglia, astrocytes and oligodendrocytes). We further investigated the potential therapeutic approaches to limit necroptosis and explained the potential mechanisms through which different drugs exert their therapeutic effects.
Further research is warranted to investigate and develop therapeutic approaches to inhibit necroptotic pathways and promote functional recovery after SCI. The RIP1-RIP3-MLKL signalling pathway plays a critical role in necroptosis, and most therapeutic options revolve around the inhibition of these three key molecules, such as Nec-1 specifically inhibiting RIP1, GSK872 inhibiting RIP3 and NSA inhibiting MLKL. It is conceivable that soon some essential molecules may be used to inhibit similar key pathways to discover new pharmacological treatments for SCI. Further investigations and more detailed clinical studies should be conducted in the future to explore the mechanisms of necroptosis in SCI and its potential role. Moreover, EA, with a traditional Chinese medicine approach, plays a non-negligible role in the treatment of SCI. Altogether, our article highlights that the inhibition of necroptosis-regulated cell death may soon represent a potential therapeutic strategy for the treatment of SCI.
Funding Statement: This review work was supported by the National Natural Science Foundation of China (Grant Nos. 81771319, 82202436); the Medical Research Project of Jiangsu Commission of Health (Grant No. ZDB2020004); the Scientific Research Project of Nantong Municipal Health Commission (Grant No. MA2021016); The First People’s Hospital of Nantong Provincial and Ministerial High-Level Science and Technology Project Cultivation Fund (Grant No. YPYJJZD009).
Author Contributions: The authors confirm their contribution to the paper as follows: study conception and design: JWF, CSW, GHX; data collection: JWF, YQL, CYJ, JLZ; draft manuscript preparation: JWF, ZMC. All authors compared different studies and approved the final version of the manuscript.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
Abarikwu SO, Pant AB, Farombi EO (2012). Dietary antioxidant, quercetin, protects sertoli-germ cell coculture from atrazine-induced oxidative damage. Journal of Biochemical and Molecular Toxicology 26: 477–485. https://doi.org/10.1002/jbt.21449 [Google Scholar] [PubMed] [CrossRef]
Al-Jundi M, Thakur S, Gubbi S, Klubo-Gwiezdzinska J (2020). Novel targeted therapies for metastatic thyroid cancer–A comprehensive review. Cancers 12: 2104. https://doi.org/10.3390/cancers12082104 [Google Scholar] [PubMed] [CrossRef]
Beattie MS, Hermann GE, Rogers RC, Bresnahan JC (2002). Cell death in models of spinal cord injury. Progress in Brain Research 137: 37–47. https://doi.org/10.1016/S0079-6123(02)37006-7 [Google Scholar] [PubMed] [CrossRef]
Bellver-Landete V, Bretheau F, Mailhot B, Vallières N, Lessard M et al. (2019). Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nature Communications 10: 518. https://doi.org/10.1038/s41467-019-08446-0 [Google Scholar] [PubMed] [CrossRef]
Boots AW, Haenen GR, Bast A (2008). Health effects of quercetin: From antioxidant to nutraceutical. European Journal of Pharmacology 585: 325–337. https://doi.org/10.1016/j.ejphar.2008.03.008 [Google Scholar] [PubMed] [CrossRef]
Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR (2006). Emerging roles of pseudokinases. Trends in Cell Biology 16: 443–452. https://doi.org/10.1016/j.tcb.2006.07.003 [Google Scholar] [PubMed] [CrossRef]
Chavez-Valdez R, Martin LJ, Flock DL, Northington FJ (2012). Necrostatin-1 attenuates mitochondrial dysfunction in neurons and astrocytes following neonatal hypoxia-ischemia. Neuroscience 219: 192–203. https://doi.org/10.1016/j.neuroscience.2012.05.002 [Google Scholar] [PubMed] [CrossRef]
Chen HC, Hsu PW, Tzaan WC, Lee AW (2014a). Effects of the combined administration of vitamins C and E on the oxidative stress status and programmed cell death pathways after experimental spinal cord injury. Spinal Cord 52: 24–28. https://doi.org/10.1038/sc.2013.140 [Google Scholar] [PubMed] [CrossRef]
Chen S, Lai W, Li X, Wang H (2022). Necrosulfonamide selectively induces DNA double-strand breaks in acute myeloid leukemia cells. Chemical Research in Toxicology 35: 387–391. https://doi.org/10.1021/acs.chemrestox.2c00044 [Google Scholar] [PubMed] [CrossRef]
Chen X, Li W, Ren J, Huang D, He WT et al. (2014b). Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Research 24: 105–121. https://doi.org/10.1038/cr.2013.171 [Google Scholar] [PubMed] [CrossRef]
Chen J, Wang S, Fu R, Zhou M, Zhang T, Pan W, Yang N, Huang Y (2018). RIP3 dependent NLRP3 inflammasome activation is implicated in acute lung injury in mice. Journal of Translational Medicine 16: 233. https://doi.org/10.1186/s12967-018-1606-4 [Google Scholar] [PubMed] [CrossRef]
Christgen S, Tweedell RE, Kanneganti TD (2022). Programming inflammatory cell death for therapy. Pharmacology and Therapeutics 232: 108010. https://doi.org/10.1016/j.pharmthera.2021.108010 [Google Scholar] [PubMed] [CrossRef]
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O et al. (2008). Identification of RIP1 kinase as a specific cellular target of necrostatins. Nature Chemical Biology 4: 313–321. https://doi.org/10.1038/nchembio.83 [Google Scholar] [PubMed] [CrossRef]
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nature Chemical Biology 1: 112–119. https://doi.org/10.1038/nchembio711 [Google Scholar] [PubMed] [CrossRef]
Ding Y, Yan Q, Ruan JW, Zhang YQ, Li WJ et al. (2011). Bone marrow mesenchymal stem cells and electroacupuncture downregulate the inhibitor molecules and promote the axonal regeneration in the transected spinal cord of rats. Cell Transplantation 20: 475–491. https://doi.org/10.3727/096368910X528102 [Google Scholar] [PubMed] [CrossRef]
Duan X, Liu X, Liu N, Huang Y, Jin Z, Zhang S, Ming Z, Chen H (2020). Inhibition of keratinocyte necroptosis mediated by RIPK1/RIPK3/MLKL provides a protective effect against psoriatic inflammation. Cell Death & Disease 11: 134. https://doi.org/10.1038/s41419-020-2328-0 [Google Scholar] [PubMed] [CrossRef]
Dunai Z, Bauer PI, Mihalik R (2011). Necroptosis: Biochemical, physiological and pathological aspects. Pathology Oncology Research 17: 791–800. https://doi.org/10.1007/s12253-011-9433-4 [Google Scholar] [PubMed] [CrossRef]
Fan H, Tang HB, Kang J, Shan L, Song H, Zhu K, Wang J, Ju G, Wang YZ (2015). Involvement of endoplasmic reticulum stress in the necroptosis of microglia/macrophages after spinal cord injury. Neuroscience 311: 362–373. https://doi.org/10.1016/j.neuroscience.2015.10.049 [Google Scholar] [PubMed] [CrossRef]
Fan H, Tang HB, Shan LQ, Liu SC, Huang DG et al. (2019). Quercetin prevents necroptosis of oligodendrocytes by inhibiting macrophages/microglia polarization to M1 phenotype after spinal cord injury in rats. Journal of Neuroinflammation 16: 206. https://doi.org/10.1186/s12974-019-1613-2 [Google Scholar] [PubMed] [CrossRef]
Fan H, Zhang K, Shan L, Kuang F, Chen K, Zhu K, Ma H, Ju G, Wang YZ (2016). Reactive astrocytes undergo M1 microglia/macrohpages-induced necroptosis in spinal cord injury. Molecular Neurodegeneration 11: 14. https://doi.org/10.1186/s13024-016-0081-8 [Google Scholar] [PubMed] [CrossRef]
Festoff BW (2014). Designing drugs that encourage spinal cord injury healing. Expert Opinion on Drug Discovery 9: 1151–1165. https://doi.org/10.1517/17460441.2014.941350 [Google Scholar] [PubMed] [CrossRef]
Fiani B, Kondilis A, Soula M, Tao A, Alvi MA (2021). Novel methods of necroptosis inhibition for spinal cord injury using translational research to limit secondary injury and enhance endogenous repair and regeneration. Neurospine 18: 261–270. https://doi.org/10.14245/ns.2040722.361 [Google Scholar] [PubMed] [CrossRef]
Fulda S (2013). The mechanism of necroptosis in normal and cancer cells. Cancer Biology & Therapy 14: 999–1004. https://doi.org/10.4161/cbt.26428 [Google Scholar] [PubMed] [CrossRef]
Gong Y, Fan Z, Luo G, Yang C, Huang Q et al. (2019). The role of necroptosis in cancer biology and therapy. Molecular Cancer 18: 100. https://doi.org/10.1186/s12943-019-1029-8 [Google Scholar] [PubMed] [CrossRef]
Grootjans S, Vanden Berghe T, Vandenabeele P (2017). Initiation and execution mechanisms of necroptosis: An overview. Cell Death and Differentiation 24: 1184–1195. https://doi.org/10.1038/cdd.2017.65 [Google Scholar] [PubMed] [CrossRef]
Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo J, Hu X (2007). Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Molecular Cancer Therapeutics 6: 1641–1649. https://doi.org/10.1158/1535-7163.MCT-06-0511 [Google Scholar] [PubMed] [CrossRef]
Hanna A, Frangogiannis NG (2019). The role of the TGF-β superfamily in myocardial infarction. Frontiers in Cardiovascular Medicine 6: 140. https://doi.org/10.3389/fcvm.2019.00140 [Google Scholar] [PubMed] [CrossRef]
He S, Liang Y, Shao F, Wang X (2011). Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proceedings of the National Academy of Sciences of the United States of America 108: 20054–20059. https://doi.org/10.1073/pnas.1116302108 [Google Scholar] [PubMed] [CrossRef]
Heiman A, Pallottie A, Heary RF, Elkabes S (2014). Toll-like receptors in central nervous system injury and disease: A focus on the spinal cord. Brain, Behavior, and Immunity 42: 232–245. https://doi.org/10.1016/j.bbi.2014.06.203 [Google Scholar] [PubMed] [CrossRef]
Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK et al. (2014). Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proceedings of the National Academy of Sciences of the United States of America 111: 15072–15077. https://doi.org/10.1073/pnas.1408987111 [Google Scholar] [PubMed] [CrossRef]
Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J (2008). Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135: 1311–1323. https://doi.org/10.1016/j.cell.2008.10.044 [Google Scholar] [PubMed] [CrossRef]
Hogan S, Canning C, Sun S, Sun X, Zhou K (2010). Effects of grape pomace antioxidant extract on oxidative stress and inflammation in diet induced obese mice. Journal of Agricultural and Food Chemistry 58: 11250–11256. https://doi.org/10.1021/jf102759e [Google Scholar] [PubMed] [CrossRef]
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nature Immunology 1: 489–495. https://doi.org/10.1038/82732 [Google Scholar] [PubMed] [CrossRef]
Hongna Y, Hongzhao T, Quan L, Delin F, Guijun L, Xiaolin L, Fulin G, Zhongren S (2020). Jia-Ji electro-acupuncture improves locomotor function with spinal cord injury by regulation of autophagy flux and inhibition of necroptosis. Frontiers in Neuroscience 14: 616864. https://doi.org/10.3389/fnins.2020.616864 [Google Scholar] [PubMed] [CrossRef]
Huang SF, Ding Y, Ruan JW, Zhang W, Wu JL, He B, Zhang YJ, Li Y, Zeng YS (2011). An experimental electro-acupuncture study in treatment of the rat demyelinated spinal cord injury induced by ethidium bromide. Neuroscience Research 70: 294–304. https://doi.org/10.1016/j.neures.2011.03.010 [Google Scholar] [PubMed] [CrossRef]
Hudobenko J, Ganesh BP, Jiang J, Mohan EC, Lee S et al. (2020). Growth differentiation factor-11 supplementation improves survival and promotes recovery after ischemic stroke in aged mice. Aging 12: 8049–8066. https://doi.org/10.18632/aging.103122 [Google Scholar] [PubMed] [CrossRef]
Ihalainen T, Rinta-Kiikka I, Luoto TM, Koskinen EA, Korpijaakko-Huuhka AM, Ronkainen A (2017). Traumatic cervical spinal cord injury: A prospective clinical study of laryngeal penetration and aspiration. Spinal Cord 55: 979–984. https://doi.org/10.1038/sc.2017.71 [Google Scholar] [PubMed] [CrossRef]
Jiang W, Huang Y, Han N, He F, Li M, Bian Z, Liu J, Sun T, Zhu L (2016). Quercetin suppresses NLRP3 inflammasome activation and attenuates histopathology in a rat model of spinal cord injury. Spinal Cord 54: 592–596. https://doi.org/10.1038/sc.2015.227 [Google Scholar] [PubMed] [CrossRef]
Jiao J, Wang Y, Ren P, Sun S, Wu M (2019). Necrosulfonamide ameliorates neurological impairment in spinal cord injury by improving antioxidative capacity. Frontiers in Pharmacology 10: 1538. https://doi.org/10.3389/fphar.2019.01538 [Google Scholar] [PubMed] [CrossRef]
Ju G, Wang J, Wang Y, Zhao X (2014). Spinal cord contusion. Neural Regeneration Research 9: 789–794. https://doi.org/10.4103/1673-5374.131591 [Google Scholar] [PubMed] [CrossRef]
Kam AE, Masood A, Shroff RT (2021). Current and emerging therapies for advanced biliary tract cancers. The Lancet Gastroenterology & Hepatology 6: 956–969. https://doi.org/10.1016/S2468-1253(21)00171-0 [Google Scholar] [PubMed] [CrossRef]
Kanno H, Ozawa H, Tateda S, Yahata K, Itoi E (2015). Upregulation of the receptor-interacting protein 3 expression and involvement in neural tissue damage after spinal cord injury in mice. BMC Neuroscience 16: 62. https://doi.org/10.1186/s12868-015-0204-0 [Google Scholar] [PubMed] [CrossRef]
Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG (2009). Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. Journal of Neuroscience 29: 13435–13444. https://doi.org/10.1523/JNEUROSCI.3257-09.2009 [Google Scholar] [PubMed] [CrossRef]
Kigerl KA, Lai W, Rivest S, Hart RP, Satoskar AR, Popovich PG (2007). Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. Journal of Neurochemistry 102: 37–50. https://doi.org/10.1111/j.1471-4159.2007.04524.x [Google Scholar] [PubMed] [CrossRef]
Lalaoui N, Lindqvist LM, Sandow JJ, Ekert PG (2015). The molecular relationships between apoptosis, autophagy and necroptosis. Seminars in Cell and Developmental Biology 39: 63–69. https://doi.org/10.1016/j.semcdb.2015.02.003 [Google Scholar] [PubMed] [CrossRef]
Li JX, Feng JM, Wang Y, Li XH, Chen XX et al. (2014). The B-RafV600E inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death & Disease 5: e1278. https://doi.org/10.1038/cddis.2014.241 [Google Scholar] [PubMed] [CrossRef]
Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K et al. (2012). The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150: 339–350. https://doi.org/10.1016/j.cell.2012.06.019 [Google Scholar] [PubMed] [CrossRef]
Liu JP (2006). The function of growth/differentiation factor 11 (Gdf11) in rostrocaudal patterning of the developing spinal cord. Development 133: 2865–2874. https://doi.org/10.1242/dev.02478 [Google Scholar] [PubMed] [CrossRef]
Liu S, Li Y, Choi HMC, Sarkar C, Koh EY, Wu J, Lipinski MM (2018). Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death & Disease 9: 476. https://doi.org/10.1038/s41419-018-0469-1 [Google Scholar] [PubMed] [CrossRef]
Lorusso L, Cappagli V, Valerio L, Giani C, Viola D et al. (2021). Thyroid cancers: From surgery to current and future systemic therapies through their molecular identities. International Journal of Molecular Sciences 22: 3117. https://doi.org/10.3390/ijms22063117 [Google Scholar] [PubMed] [CrossRef]
McPherron AC, Lawler AM, Lee SJ (1999). Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nature Genetics 22: 260–264. https://doi.org/10.1038/10320 [Google Scholar] [PubMed] [CrossRef]
Mehta SL, Manhas N, Raghubir R (2007). Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Research Reviews 54: 34–66. https://doi.org/10.1016/j.brainresrev.2006.11.003 [Google Scholar] [PubMed] [CrossRef]
Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM et al. (2013). M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nature Neuroscience 16: 1211–1218. https://doi.org/10.1038/nn.3469 [Google Scholar] [PubMed] [CrossRef]
Motawi TMK, Abdel-Nasser ZM, Shahin NN (2020). Ameliorative effect of necrosulfonamide in a rat model of alzheimer’s disease: Targeting mixed lineage kinase domain-like protein-mediated necroptosis. ACS Chemical Neuroscience 11: 3386–3397. https://doi.org/10.1021/acschemneuro.0c00516 [Google Scholar] [PubMed] [CrossRef]
O’Shea TM, Burda JE, Sofroniew MV (2017). Cell biology of spinal cord injury and repair. Journal of Clinical Investigation 127: 3259–3270. https://doi.org/10.1172/JCI90608 [Google Scholar] [PubMed] [CrossRef]
Ransohoff RM, Brown MA (2012). Innate immunity in the central nervous system. Journal of Clinical Investigation 122: 1164–1171. https://doi.org/10.1172/JCI58644 [Google Scholar] [PubMed] [CrossRef]
Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P et al. (2016). Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death and Differentiation 23: 76–88. https://doi.org/10.1038/cdd.2015.70 [Google Scholar] [PubMed] [CrossRef]
Schwab JM, Zhang Y, Kopp MA, Brommer B, Popovich PG (2014). The paradox of chronic neuroinflammation, systemic immune suppression, autoimmunity after traumatic chronic spinal cord injury. Experimental Neurology 258: 121–129. https://doi.org/10.1016/j.expneurol.2014.04.023 [Google Scholar] [PubMed] [CrossRef]
Shan B, Pan H, Najafov A, Yuan J (2018). Necroptosis in development and diseases. Genes and Development 32: 327–340. https://doi.org/10.1101/gad.312561.118 [Google Scholar] [PubMed] [CrossRef]
Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM (2007). Necrostatin: A potentially novel cardioprotective agent? Cardiovascular Drugs and Therapy 21: 227–233. https://doi.org/10.1007/s10557-007-6035-1 [Google Scholar] [PubMed] [CrossRef]
Sugaya T, Kanno H, Matsuda M, Handa K, Tateda S, Murakami T, Ozawa H, Itoi E (2019). B-RAFV600E inhibitor dabrafenib attenuates RIPK3-mediated necroptosis and promotes functional recovery after spinal cord injury. Cells 8: 1582. https://doi.org/10.3390/cells8121582 [Google Scholar] [PubMed] [CrossRef]
Sun L, Wang H, Wang Z, He S, Chen S et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148: 213–227. https://doi.org/10.1016/j.cell.2011.11.031 [Google Scholar] [PubMed] [CrossRef]
Sutherland BA, Hadley G, Alexopoulou Z, Lodge TA, Neuhaus AA, Couch Y, Kalajian N, Morten KJ, Buchan AM (2020). Growth differentiation factor-11 causes neurotoxicity during ischemia in vitro. Frontiers in Neurology 11: 1023. https://doi.org/10.3389/fneur.2020.01023 [Google Scholar] [PubMed] [CrossRef]
Wang Y, Jiao J, Zhang S, Zheng C, Wu M (2019). RIP3 inhibition protects locomotion function through ameliorating mitochondrial antioxidative capacity after spinal cord injury. Biomedicine and Pharmacotherapy 116: 109019. https://doi.org/10.1016/j.biopha.2019.109019 [Google Scholar] [PubMed] [CrossRef]
Wang Y, Wang H, Tao Y, Zhang S, Wang J, Feng X (2014). Necroptosis inhibitor necrostatin-1 promotes cell protection and physiological function in traumatic spinal cord injury. Neuroscience 266: 91–101. https://doi.org/10.1016/j.neuroscience.2014.02.007 [Google Scholar] [PubMed] [CrossRef]
Wang Y, Wang J, Wang H, Feng X, Tao Y, Yang J, Cai J (2018). Necrosulfonamide attenuates spinal cord injury via necroptosis inhibition. World Neurosurgery 114: e1186–e1191. https://doi.org/10.1016/j.wneu.2018.03.174 [Google Scholar] [PubMed] [CrossRef]
Wang Y, Wang J, Yang H, Zhou J, Feng X, Wang H, Tao Y (2015). Necrostatin-1 mitigates mitochondrial dysfunction post-spinal cord injury. Neuroscience 289: 224–232. https://doi.org/10.1016/j.neuroscience.2014.12.061 [Google Scholar] [PubMed] [CrossRef]
Wang S, Wu J, Zeng YZ, Wu SS, Deng GR, Chen ZD, Lin B (2017). Necrostatin-1 mitigates endoplasmic reticulum stress after spinal cord injury. Neurochemical Research 42: 3548–3558. https://doi.org/10.1007/s11064-017-2402-x [Google Scholar] [PubMed] [CrossRef]
Wu C, Chen J, Liu Y, Zhang J, Ding W et al. (2016). Upregulation of PSMB4 is associated with the necroptosis after spinal cord injury. Neurochemical Research 41: 3103–3112. https://doi.org/10.1007/s11064-016-2033-7 [Google Scholar] [PubMed] [CrossRef]
Xu X, Chua KW, Chua CC, Liu CF, Hamdy RC, Chua BH (2010). Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain Research 1355: 189–194. https://doi.org/10.1016/j.brainres.2010.07.080 [Google Scholar] [PubMed] [CrossRef]
Xu Y, Hu X, Li F, Zhang H, Lou J et al. (2021). GDF-11 protects the traumatically injured spinal cord by suppressing pyroptosis and necroptosis via TFE3-mediated autophagy augmentation. Oxidative Medicine and Cellular Longevity 2021: 8186877. https://doi.org/10.1155/2021/8186877 [Google Scholar] [PubMed] [CrossRef]
Xu YJ, Zheng L, Hu YW, Wang Q (2018). Pyroptosis and its relationship to atherosclerosis. Clinica Chimica Acta 476: 28–37. https://doi.org/10.1016/j.cca.2017.11.005 [Google Scholar] [PubMed] [CrossRef]
Ying Y, Padanilam BJ (2016). Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis? Cellular and Molecular Life Sciences 73: 2309–2324. https://doi.org/10.1007/s00018-016-2202-5 [Google Scholar] [PubMed] [CrossRef]
Yu Z, Jiang N, Su W, Zhuo Y (2021). Necroptosis: A novel pathway in neuroinflammation. Frontiers in Pharmacology 12: 701564. https://doi.org/10.3389/fphar.2021.701564 [Google Scholar] [PubMed] [CrossRef]
Zha J, Smith A, Andreansky S, Bracchi-Ricard V, Bethea JR (2014). Chronic thoracic spinal cord injury impairs CD8+ T-cell function by up-regulating programmed cell death-1 expression. Journal of Neuroinflammation 11: 65. https://doi.org/10.1186/1742-2094-11-65 [Google Scholar] [PubMed] [CrossRef]
Zhang QX, Guo D, Wang FC, Ding WY (2020). Necrosulfonamide (NSA) protects intervertebral disc degeneration via necroptosis and apoptosis inhibition. European Review for Medical and Pharmacological Sciences 24: 2683–2691. https://doi.org/10.26355/eurrev_202003_20538 [Google Scholar] [PubMed] [CrossRef]
Zhao Y, Wang LH, Peng A, Liu XY, Wang Y, Huang SH, Liu T, Wang XJ, Chen ZY (2020). The neuroprotective and neurorestorative effects of growth differentiation factor 11 in cerebral ischemic injury. Brain Research 1737: 146802. https://doi.org/10.1016/j.brainres.2020.146802 [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools