
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Role of RIPK1 in the pathogenesis of acute respiratory distress syndrome
School of Environmental and Biological Engineering, Nanjing University of Science & Technology, Nanjing, China
* Corresponding Author: DAN WENG. Email:
BIOCELL 2023, 47(10), 2151-2162. https://doi.org/10.32604/biocell.2023.030570
Received 13 April 2023; Accepted 26 July 2023; Issue published 08 November 2023
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Acute respiratory distress syndrome (ARDS) is a life-threatening pulmonary disease typically caused by microbial infections, trauma, inhalation of harmful gases, and other factors. It is characterized by an inflammation in the lungs and increased alveolar permeability, leading to pulmonary edema and consequently, a low oxygen supply or hypoxemia. ARDS is responsible for 1 in 10 admissions to intensive care units, and the mortality rate for patients with severe ARDS is as high as 46%. Extensive efforts have been devoted to investigating the pathological mechanisms of ARDS to develop new effective clinical strategies. Recent studies have reported that receptor-interacting serine/threonine kinase 1 (RIPK1) is involved in the pathogenesis of ARDS. RIPK1 is a critical mediator of programmed cell death and inflammation. Growing evidence suggests that RIPK1 plays a role in the pathogenesis of different inflammatory diseases and serves as a promising pharmaceutical target. This review summarizes and sheds some light on the recent findings regarding the role of RIPK1 and related molecules in the pathogenesis of ARDS.Graphic Abstract
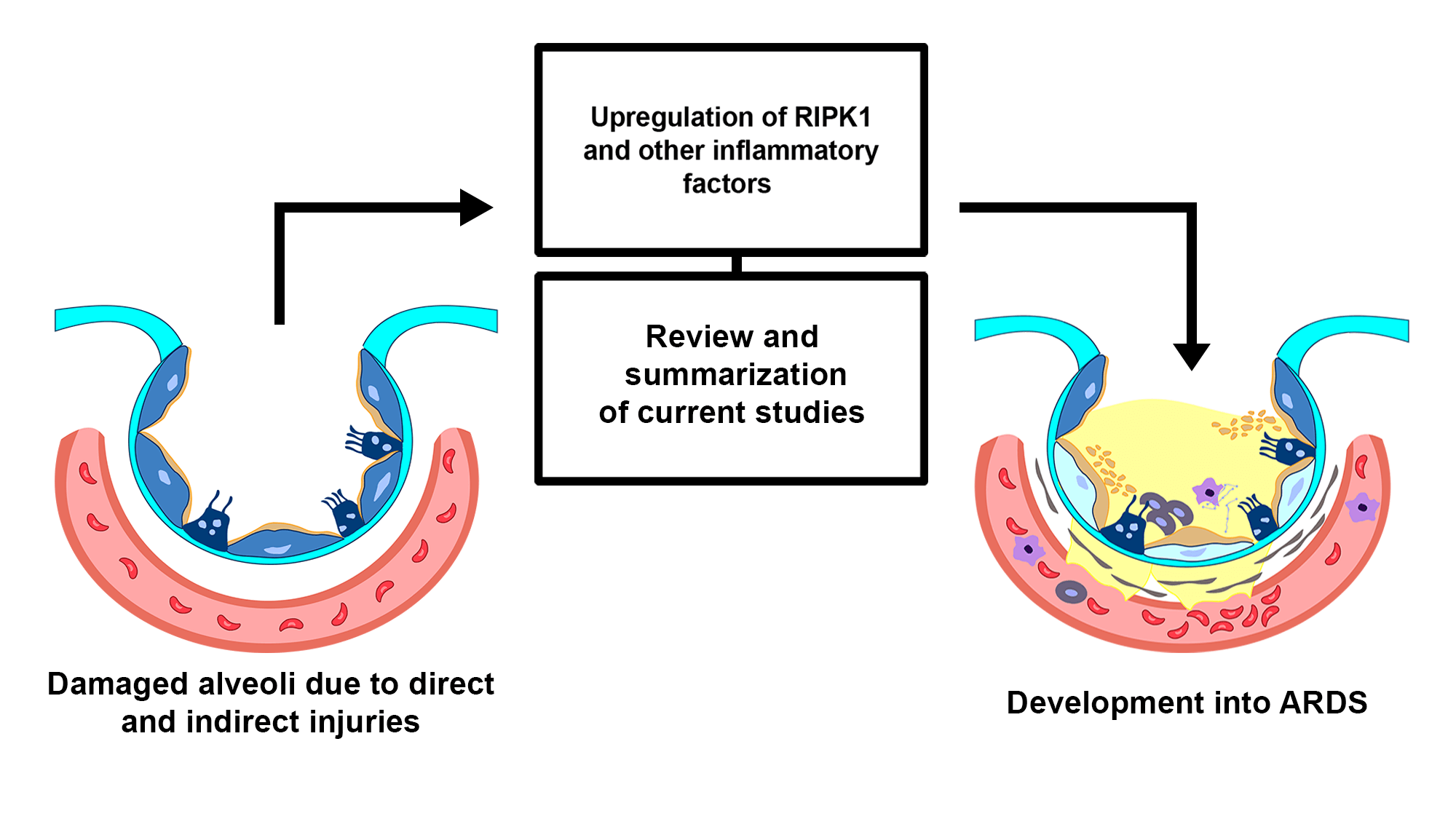
Keywords
Acute respiratory distress syndrome (ARDS), a severe form of acute lung injury (ALI), is a medical condition arising from a lung injury and subsequent inflammatory response, characterized by pulmonary edema/hyper-permeability and hypoxemia. It is known to occur after exposure to infection, trauma, inhalation, and other stimuli. Pulmonary inflammatory factors accumulate in this condition, and at the same time, the pulmonary capillaries are damaged, the permeability is altered, and tissue fluid enters the alveoli, leading to the formation of pulmonary dead space. The accumulation of hyaline membrane (pulmonary edema) can disrupt the gas exchange between the alveoli and capillary, causing low oxygen supply (hypoxemia). This can consequently lead to severe shortness of breath, weakness, cough, fever, and collapsed alveoli. On the basis of the decreased blood pressure, ADRS is measured by the ratio of arterial oxygen partial pressure to fractional inspired oxygen (PaO2/FiO2). The ratio is categorized by the 2012 Berlin conference to identify the magnitude of ARDS and to further determine its underlying causes as mild (200–300 mmHg), moderate (100–200 mmHg), or severe (<100 mmHg) levels (Ranieri et al., 2012; Papazian et al., 2019; He et al., 2021). Moreover, previous studies have shown a definite involvement of several signaling pathways, such as the nuclear factor-κB (NF-κB) pathway and the mediating role of RIPK1, in conjunction with the development of ARDS. Currently, the underlying mechanism of ARDS has been the subject of extensive research. However, the specific role of RIPK1 in it has not been fully elucidated, hence, clarifying the role of RIPK1 in ARDS may provide some insights for future studies.
Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) is a member of the serine/threonine/tyrosine kinase family that mediates the innate immune cell defense in response to various injuries as well as up-regulating the inflammatory genes, this multi-domain protein kinase determines whether a cell would undergo a pro-survival pathway (NF-κB) or the programmed cell death pathway (necroptosis or apoptosis) (Ofengeim and Yuan, 2013; Degterev et al., 2014; Cuny and Degterev, 2021). The scaffolding function and role of RIPK1 in kinase-mediated cell death have been studied with much emphasis. Further, this protein kinase was proposed to be the link to several pathways for apoptosis, necroptosis, and pyroptosis, with the first two pathways being heavily influenced by RIPK1 (D’Arcy, 2019; Malireddi et al., 2020). RIPK1 contains a C-terminal death domain which is an important binding mediator to specific death receptor signals during tumor necrosis factor-alpha (TNF-α) stimulation. Its N-terminal serine/threonine kinase domain is responsible for the necroptosis pathway and the RIPK1-dependent apoptosis pathway. Additionally, the intermediate domain mediates the activation of the pro-survival NF-κB and mitogen-activated protein kinase (MAPK) via K377 ubiquitination by cellular inhibitor of apoptosis (cIAP1/2) (Annibaldi et al., 2018) and binding ligases (Meng et al., 2018). Interestingly, RIPK1 can mediate the necroptosis and apoptosis pathways by forming several complexes depending on the degree of inflammation (Degterev et al., 2014; Liu et al., 2021). These types of regulated cell death pathways, especially based on the activation of RIPK1, are well-known in countless inflammatory diseases such as multiple sclerosis and other neurodegenerative diseases (Yuan et al., 2019). Since ARDS is described as an inflammatory disease prompted by cytokine storm and alveolus damage, it is evident that RIPK1 is one of the many vital molecules taking part in this inflammatory process.
Acute Respiratory Distress Syndrome (ARDS): An Overview
Early pathological stages of acute respiratory distress syndrome
The pathogenesis of ARDS involves a complex process of lung injury and inflammation that results in impaired gas exchange and respiratory failure. It can be divided into three phases: exudative, proliferative, and fibrosis phases. This review will focus on the exudative phase since it deals with the preliminary stage characterized by excessive inflammation, hyaline membrane formation, and hypoxemia (Walkey et al., 2012). The exudative phase can be further broken down into six sub-stages to better comprehend the pathological features and mechanisms.
In the first stage (Fig. 1), the lungs are initially damaged following exposure to external trauma, aspiration, infection, or injury. The alveolar blood flow then decreases, platelets begin to aggregate, and immune cells begin to release inflammatory mediators such as histamine, serotonin, and leukotrienes. This further recruits other immune cells and the release of pro-inflammatory cytokines including, tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1β, and IL-6 that initiates several signaling pathways (Borriello et al., 2017; Kanaoka and Austen, 2019; Karhausen et al., 2020; Liu et al., 2022). In the second stage (Fig. 1), these inflammatory mediators damage the alveolar-capillary membrane, increasing its permeability, and causing fluid to shift into the tissue space to result in tissue edema (Matthay et al., 2012). At the same time, the released inflammatory mediators and activated immune cells can also lead to the recruitment of neutrophils and macrophages, which further contribute to lung injury and inflammation. Neutrophils release toxic substances, including neutrophil extracellular trap (NET), that damage the lung tissue, while macrophages release cytokines that attract more immune cells to the site of injury (Williams et al., 2017; Abdulnour et al., 2018; Herrera et al., 2022). In stages 3 and 4 (Fig. 2), as lung tissue damage worsens, capillary permeability further increases, leading to protein and fluid exudation and a spike in tissue interstitial fluid osmotic pressure, resulting in pulmonary edema formation. This consequently impairs the lung capillary function, including a decrease in lung blood flow, destruction of surface-active substances due to alveolar fluid, alveolar collapse, and occurrence of atelectasis which limits the gas exchange, hence a decrease in lung compliance (Matthay et al., 2012; Mokra, 2020). In stage 5 (Fig. 3), even with adequate oxygen, the oxygen cannot enter the blood, but CO2 can still be released, leading to decreased oxygen supply in the blood with a PaO2/FiO2 that is less than 300 mmHg, and the pulmonary edema continues to worsen (Ohshimo, 2021). In stage 6 (Fig. 3), excessive inflammation leads to pulmonary tissue fibrosis, diffuse disorders worsen, and lung tissue becomes stiffer and less compliant, leading to long-term respiratory impairment (Gorman et al., 2022). Besides, stage 6 also presents CO2 retention lowering the pH level of the patient and adding more deleterious outcomes for ARDS (Peschel et al., 2021; Maamar et al., 2023).
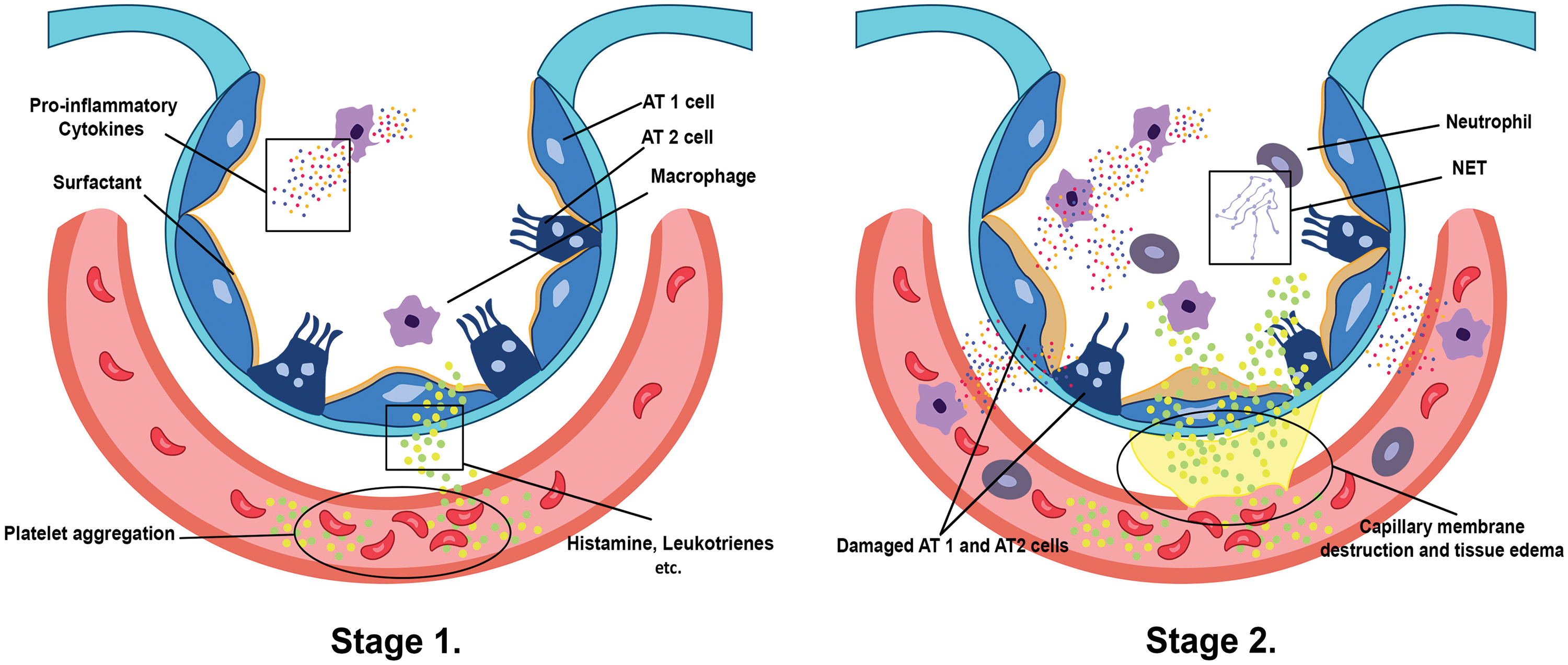
Figure 1: Stage 1 presents how platelet aggregation triggers mast cells, basophils, and others to release histamine, leukotrienes, and several inflammatory mediators in the interstitium, which recruit/promote other immune cells, such as the alveolar macrophages, to release pro-inflammatory cytokines (Borriello et al., 2017; Kanaoka and Austen, 2019; Karhausen et al., 2020). The inflammatory mediators and cytokines inflict damage to alveolar epithelium type 1/2 (AT1/2) cells as well as the capillary membrane, which signals a rush of neutrophils and several immune cells to the affected alveoli and site of platelet aggregation. Additionally, neutrophils secrete neutrophil extracellular traps (NETs) inside the alveolar space and capillary, wreaking havoc on the lung tissue and contributing to the formation of tissue edema as shown in stage 2 (Matthay et al., 2012; Williams et al., 2017; Abdulnour et al., 2018; Herrera et al., 2022).
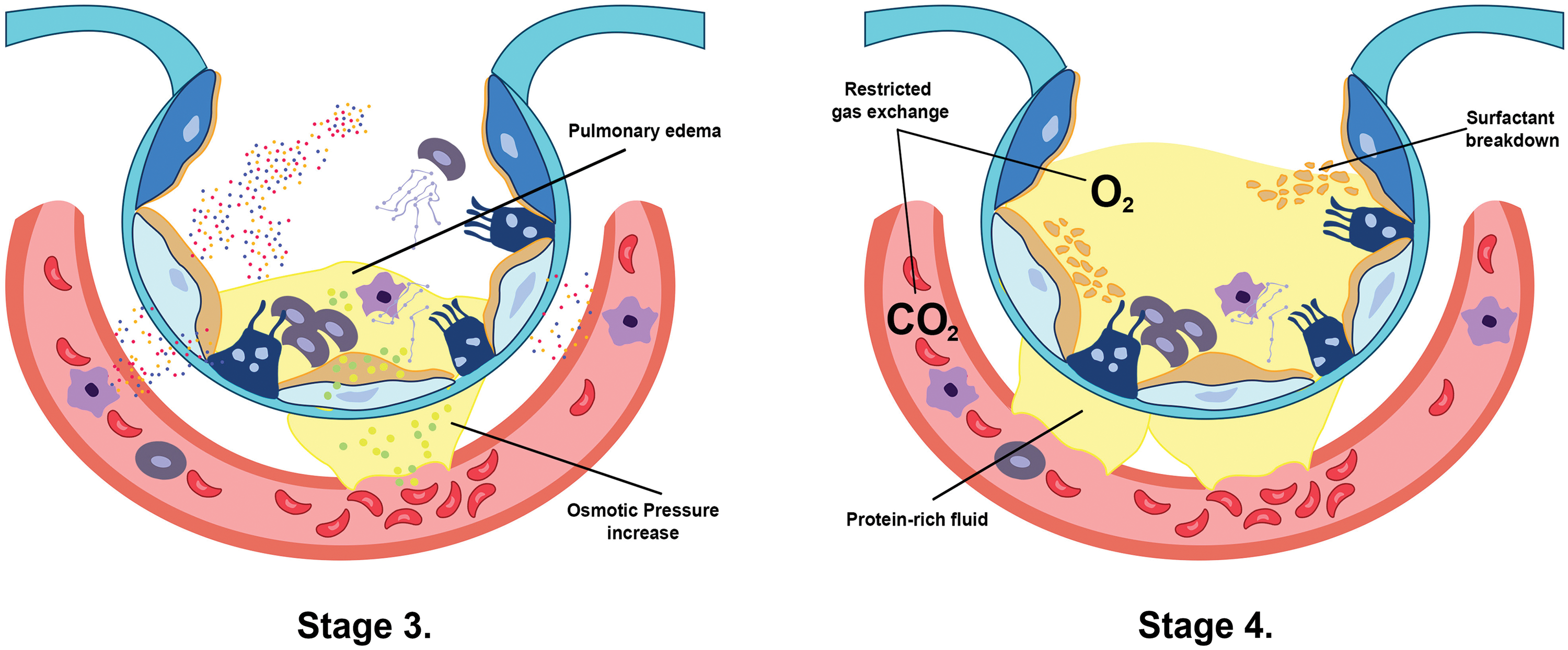
Figure 2: The increase of capillary membrane permeability in stage 3 creates an osmotic pressure between the interstitial spaces due to fluid shift, which induces pulmonary edema formation (Matthay et al., 2012). Furthermore, this protein-rich edema breaks down the surfactant, leading to alveolar collapse and limiting the exchange between oxygen and carbon dioxide in stage 4 (Mokra, 2020).
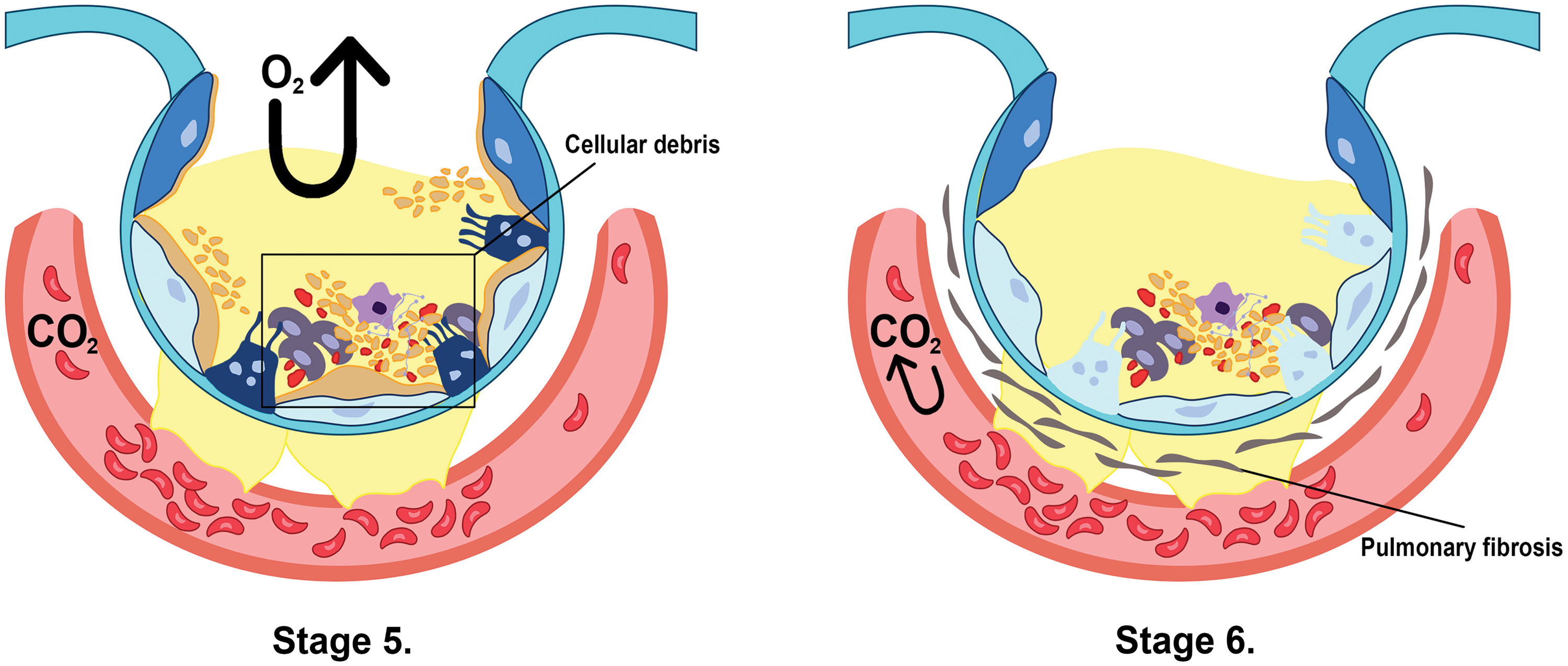
Figure 3: The combination of cellular debris in the fluid and atelectasis renders it impossible for the oxygen to get into the capillary membrane in stage 5. In contrast, stage 6 shows pulmonary tissue fibrosis, which indefinitely restricts the gas exchange between the alveoli and the membrane, as well as carbon dioxide retention in the blood (Ohshimo, 2021; Peschel et al., 2021; Gorman et al., 2022; Maamar et al., 2023).
Overall, the pathogenesis of ARDS involves a complex interplay between the immune system, inflammatory mediators, and lung tissue damage, resulting in impaired gas exchange and respiratory failure. From a radiological perspective, the pathogenesis of ARDS is characterized by bilateral and patchy ground-glass opacities in the early exudative phase, which corresponds to interstitial edema and hyaline membranes. The spatial distribution of patchy ground-glass opacities and areas of lobular sparing and basal consolidations are the characteristic imaging features of ARDS (Meyer et al., 2021). In the proliferative and fibrotic phases of the disease, the radiological features are characterized by traction bronchiectasis or bronchiectasis within areas of increased attenuation on high-resolution computed tomography (Butt et al., 2016).
Main Factors Promoting the Development of ARDS
ARDS is a severe respiratory condition that occurs when fluid leaks into the lung’s air sacs, resulting in respiratory failure. Current studies state that there are several pathological mechanisms that contribute to the development of ARDS.
For instance, direct lung injury, such as pneumonia (viral and bacterial), mechanical injury, or inhalation of gastric contents, can cause damage to the alveolar-capillary membrane, leading to inflammation, edema, and impaired gas exchange (D’Alessio, 2018). Alternatively, indirect lung injury is caused by non-pulmonary factors, mainly sepsis, trauma, or pancreatitis. These injuries can lead to systemic inflammation, which can then cause damage to the alveolar-capillary membrane and result in ARDS (Mokra, 2020).
Both direct and indirect injuries mentioned above systematically induce an inflammatory response and release inflammatory mediators, such as cytokines, chemokines, and leukotrienes during the early exudative phase of ARDS. One of the pathways responsible for ARDS inflammation is the NF-κB signaling pathway. The TNF-α cytokine induces its activation, stimulating macrophages to release IL-6, IL-2, and other pro-inflammatory cytokines. Additionally, under the TNF-α stimulation, cell death such as necroptosis and RIPK1-dependent apoptosis, can occur (Yuan et al., 2019; Li et al., 2022). Another form of cell death causing damage to the alveolar cell barrier is pyroptosis, known for its caspase activation and gasdermin D (GSDMD) cleavage. The aspects of osmotic imbalance and the release of IL-1β and IL-18 could bring devastating effects such as continuous recruitment of immune cells and damage-associated molecular patterns (DAMPs) release that includes the protein high-mobility group box 1 (HMGB1) and lactate dehydrogenase (LDH) (Roh and Sohn, 2018; Tan et al., 2021; Zheng et al., 2023). The accumulation of reactive oxygen species (ROS) and iron accumulation, along with lipid peroxidation are the main features of ferroptosis. Just like immunogenic cell death, it further recruits immune cells to the site of damage, and much like pyroptosis, it also contributes to the release of pro-inflammatory cytokines (Zheng et al., 2023). These mediators cause major damage to alveolar epithelial cells that line the air sacs in the lungs, consequently disrupting the alveolar-capillary membrane. This further leads to increased permeability and fluid accumulation in the lungs and the subsequent development of pulmonary edema. Likewise, injury to the endothelial cells, which line the blood vessels in the lungs, can also lead to disruption of the alveolar-capillary membrane, adding to the fluid accumulation (Chen et al., 2021b). Many times such damage to lung cells could also result in microthrombi formation, making patients with ARDS susceptible to coagulation abnormalities, causing additional damage to the alveolar-capillary membrane and worsening the condition (Mackman et al., 2020). Since ARDS is a complex inflammatory disease involving multiple pathological mechanisms, these factors and cell death pathways significantly contribute to the development of respiratory failure.
Characteristics of Acute Respiratory Distress Syndrome
Development of pulmonary edema
At present, the pathogenesis of ARDS mainly revolves around the occurrence and regulation of pulmonary edema and inflammation, with pulmonary edema deemed an important clinical feature of ALI and ARDS (Herrero et al., 2018). Currently, there are two generally recognized reasons for pulmonary edema in ALI/ARDS. The first is the damage to pulmonary microvascular endothelial cells, which increases the permeability of local capillaries. Further, the promotion of the exudation of plasma-rich protein leads to the “swelling” of tissue water. The second is the abnormal quantity and function of aquaporins (Saguil and Fargo, 2020; Zeng et al., 2021). Alveolar fluid is cleared through active or passive transport, in which active transport is dominant. The aquaporin is an important medium for water exchange between capillaries and air (Papadopoulos et al., 2008; Rahmel et al., 2018). In mammals, 13 aquaporins have been documented of which 6 aquaporins are distributed in the lung tissue (Vassiliou et al., 2017). Studies have shown that the expression and function of aquaporins are affected due to lung diseases, and the fluid clearance in the alveoli decreases, resulting in pulmonary edema (Xie et al., 2005; Yadav et al., 2020). The accumulation of edema fluid in the pulmonary interstitium and interstitial space will increase the work done by the respiratory exchange. This inhibition of gas exchange, reduction of oxygen intake as well as carbon dioxide excretion, further leads to the development of hypoxia into acute respiratory failure (Liu et al., 2022).
Inflammation and immune cell infiltration
In earlier studies, ALI/ARDS was found to be a complex and excessive inflammatory response of lung tissue after external or internal stimulation through several signaling pathways, including NF-κB, Janus kinases 2/signal transducers and activators of transcription 3 (JAK2/STAT3), and MAPK to name a few. Hence, it is of great significance to study the pathogenesis of inflammation in order to reveal the pathogenesis of ALI/ARDS (Li et al., 2022). A dysfunctional inflammatory response initially drives ARDS with a rise in the number of inflammatory cells after lung injury, including polymorphonuclear leukocytes (PMN), monocytes, and macrophages. Further, endogenous molecules related to cell damage can recognize and bind to toll-like receptors (TLRs) on lung epithelial cells and alveolar macrophages and activate the innate immune system (Bakopoulos et al., 2017). The innate immune system defense mechanisms, such as the formation of NET and the release of histones, can help capture pathogens but may aggravate alveolar damage (Huppert et al., 2019). As the most typical inflammatory response feature in lung injury, neutrophil recruitment also plays an important role during lung damage (Potey et al., 2019). A lung tissue that has been severely injured, infected, or has been exposed to inhaled toxic gases produces a large number of inflammatory mediators and lipid metabolites. This promotes the recruitment and activation of inflammatory cells, including neutrophils and macrophages, in the lung tissue. This forms a “cell network” of inflammatory response and immune regulation in ALI/ARDS, such as the release of cytokines (IL-1 β, IL-6, IL-8, and TNF-α, etc.) and activation of the mentioned signaling pathways, thus, forming the chain reaction of inflammation (Vassiliou et al., 2017; Potey et al., 2019; Williams et al., 2017). Though to some extent, this chain reaction helps to eliminate harmful pathogens, it consequently causes damage to vascular endothelial cells and lung epithelial cells. This affects the material exchange between cells and tissues, then allowing protein-rich fluids to enter the lung alveoli, which also grounds for acute lung edema formation.
Under normal conditions, PMN plays an important role in the host’s defense against foreign stimuli. The cells aid host cells in killing microorganisms and reducing or controlling acute inflammatory response (Akgul et al., 2001). Still, at the same time, PMN release substances such as superoxide and elastase, which leads to lung tissue damage and prolong the viability of the PMN (Coldren et al., 2006). Murine animal model studies have found that selective depletion of PMN can reduce the production of tissue inflammatory factors such as IL-8, and reduce lung tissue damage (Aeffner et al., 2015; Koehler et al., 2020). However, there is a delay in the apoptosis of PMN exuded into lung tissue during the onset of ARDS. Further, alveolar lavage fluid of patients with early ARDS can inhibit neutrophil apoptosis in vitro, while this inhibition disappears when inflammation in late ARDS is eliminated (Pallister, 2005).
In the inflammatory exudative stage of ARDS, various inflammatory mediators, including leukotrienes, prostaglandins, ROS, proteases, and cytokines are formed/released. Of these, TNF and interleukins are the most influential. Research showed that TNF-α plays an important role in capillary permeability change, inducing the activation of endothelial cell activation, and the migration of leukocytes, etc. Further, it is also an important cascade-initiating factor in the inflammatory response (Pooladanda et al., 2019). The dynamic change of TNF-α is earlier than that of IL-1 β and IL-8 (Liu et al., 2022). Hence, the content of TNF-α in lung tissue or alveolar lavage fluid can be measured to determine whether ALI/ARDS occurs or not. Additionally, IL-1, IL-6, and IL-8 play an important role in the pathogenesis of ARDS, they can induce the chemotaxis of inflammatory cells such as PMN, release inflammatory mediators, and pyrogens (Song et al., 2020). It is worth noting that after the acute inflammatory stage, there will be lung tissue repair through the JAK2/STAT3 pathway as well as the proliferation of type II alveolar epithelium cells. However, its excessive activation along with the other inflammatory pathways, can lead to differentiation of type I alveolar epithelial cells, lymphocyte infiltration, and a significant secretion of surfactant (Li et al., 2022). These factors, which produce excessive cellular collagen tissue and formation of extensive fibrosis, cause a reduction in lung compliance, an increase of alveolar dead space, and stubborn hypoxemia (Paris et al., 2020; Gokey et al., 2021; Li et al., 2022).
Treatment Measures for Acute Respiratory Distress Syndrome
Given that ALI and ARDS are recognized as serious public health problems due to their high incidence and mortality rate, exploring more convenient and efficient treatment methods for ARDS has always been a hot topic in the medical science community (Dawood et al., 2012; Friedrichson et al., 2021). The general treatment for ARDS is to take respiratory support and actively treat the causes or inducing causes, including keeping the respiratory tract unobstructed, addressing hypoxia and improving ventilation, monitoring and supporting other important organ functions. Treatment is mainly divided into mechanical ventilation, drug, surgical, nutritional treatment, and other methods. Drug therapy, including glucocorticoids, anti-inflammatory drugs, respiratory stimulants, and antioxidants, has already been tested in the clinical trials of ARDS (Yang et al., 2018). Interestingly, the use of exosomes such as the mesenchymal stem cell (MSCs)-derived exosomes was seen to inhibit many pro-inflammatory cytokines and differentiation of T cells into Th17 cells in endotoxin-induced mice. Further, it also promoted cell regeneration, including the proliferation of alveolar type 2 progenitor (AT 2) (Olajuyin et al., 2019) cells and the re-epithelialization of damaged lung cells. Furthermore, in another study, microRNA-21-5p successfully reduced several pro-apoptotic genes and inhibited caspase expression (Li et al., 2015; Yin et al., 2019; Liu et al., 2022). These potential novel treatments, accompanied by traditional methods such as mechanical ventilation, may effectively enhance the survivability of ARDS patients.
Generally, ALI and ARDS patients do not need surgery, but once massive hemoptysis occurs, the airway should be kept smooth, artificial airway and endotracheal intubation should be established immediately. Double lumen endotracheal intubation is preferred, and surgery should be performed when necessary (Goh and Kong, 2020). Mechanical ventilation is the basic treatment measure to improve the gas exchange disorder of ARDS, such as optimizing the positive end-expiratory pressure or PEEP (Sahetya et al., 2017; Liaqat et al., 2022). Mechanical ventilation auxiliary treatment is an important component of the current mechanical ventilation strategy, such as prone position ventilation technology, extracorporeal membrane oxygenation technology, etc. However, this method can also bring invasive damage to the lung, which is deleterious and life-threatening (Bates and Smith, 2018).
Receptor-Interacting Serine/Threonine-Protein Kinase 1 (RIPK1): An Overview
RIPK1 is a protein kinase related to cell death and is important in various cell death pathways. RIPK1 mainly participates in multiple biological processes, such as apoptosis, necrosis, and inflammation.
The structure of RIPK1 includes the N-terminal death domain (DD), the middle conjugate enzyme structure domain (kinase-like domain, KD), and the C-terminal serine/threonine kinase domain. The DD domain can interact with other proteins containing DD domains, thereby forming a signal complex. The KD domain has kinase activity, which can auto-phosphorylate and cross-phosphorylate other target proteins and, the kinase domain is the final catalytic part of RIPK1 (Yuan et al., 2019).
The mechanism of RIPK1 in inducing inflammation or cell death
When external stimuli stimulate cells, certain pro-inflammatory cytokines released by innate immune cells provide a host-defense mechanism that conducts the cell to undertake three signaling pathways, namely NF-κB, apoptosis, or necroptosis (Qadri et al., 2018; Chen et al., 2021a). Initially, the binding of TNF-α cytokine to the trimerized receptor TNFR-1, followed by the recruitment of the C-terminal DD and the TNFR1-associated death domain protein (TRADD) of RIPK1 to the DD of the trimerized receptor, generally initiates the pro-survival or cell death regulation.
After its recruitment, the activated RIPK1, identified by the S166 phosphorylation, is subjected to a series of complex formations. Complex I begins with the binding of TRADD-RIPK1 to the TNFR-associated factor-2 (TRAF2), after which the cIAP1/2 is recruited to create K63 ubiquitin chains. This K63 chain binds with the linear ubiquitination chain assembly complex (LUBAC), which does linear ubiquitylation to TRADD-RIPK1 (Yuan et al., 2019). After the linear ubiquitylation of the TRADD-RIPK1 complex, K63 ubiquitin chains utilize and bind with TAK1 (transforming growth factor-β-activated kinase 1) and TAK1-binding protein 2/TAK1-binding protein 3 (TAB2/TAB3). Additionally, the linear ubiquitin chain activates the NF-κB essential modulator (NEMO) and IKK (IκB kinase). These two elements are important for the pro-survival NF-kβ signaling pathway, which regulates the transcriptional expression of inflammatory genes such as nucleotide-binding domain and leucine-rich repeat-containing protein 3 (NLRP3), AC20, and cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein (cFLIP) (Dolcet et al., 2005; Safa, 2013; Zhang et al., 2019).
LUBAC can be ubiquitinated through the CYLD enzyme, the inhibition of TAK1 (transforming growth factor-β-activated kinase-1) as well as the inhibitor of apoptosis protein (cIAP1 and cIAP2). The deubiquitination of LUBAC initiates the phosphorylation and dissociation of RIPK1 from complex I leading to its dimerization. Further, the splitting of this dimerized pRIPK1 is essential in RIP1-dependent apoptosis and necroptosis (Meng et al., 2018; Cao and Mu, 2021). The interaction of p-RIPK1 with RIPK3 phosphorylates this protein kinase forming the necrosomal assembly/complex IIb (pRIPK1-pRIPK3), which then drives the recruitment and oligomerization of the mixed lineage kinase domain-Like protein (pRIPK1-pRIPK3-MLKL), MLKL is then translocated to the cell membrane and triggers necroptosis (Geng et al., 2017; Sauler et al., 2019; Yuan et al., 2019). On the other hand, the formation of FADD-RIPK1-caspase 8-cFLIP (complex IIa), in which the removal of the cFLIP inhibitor of caspase 8, induces the caspase cascade (Caspase-3/7) initiating the extrinsic RIPK1-dependent apoptosis (Sauler et al., 2019). Moreover, the active caspase-8 can cleave RIPK1, further contributing to the necrosomal assembly formation (Fig. 4). Activation of RIPK1 plays a major role in these pathways, where its recruitment to the death domain of specific protein receptors and its scaffolding function helps to regulate the survival and cell death during an exogenous or endogenous injury. However, there are studies suggesting that this protein kinase could either be therapeutic (inhibition of necrostatin-1 stable or Nec-1s) or harmful (cytokine storm) (Yuan et al., 2019; Cao and Mu, 2021; Liu et al., 2023).
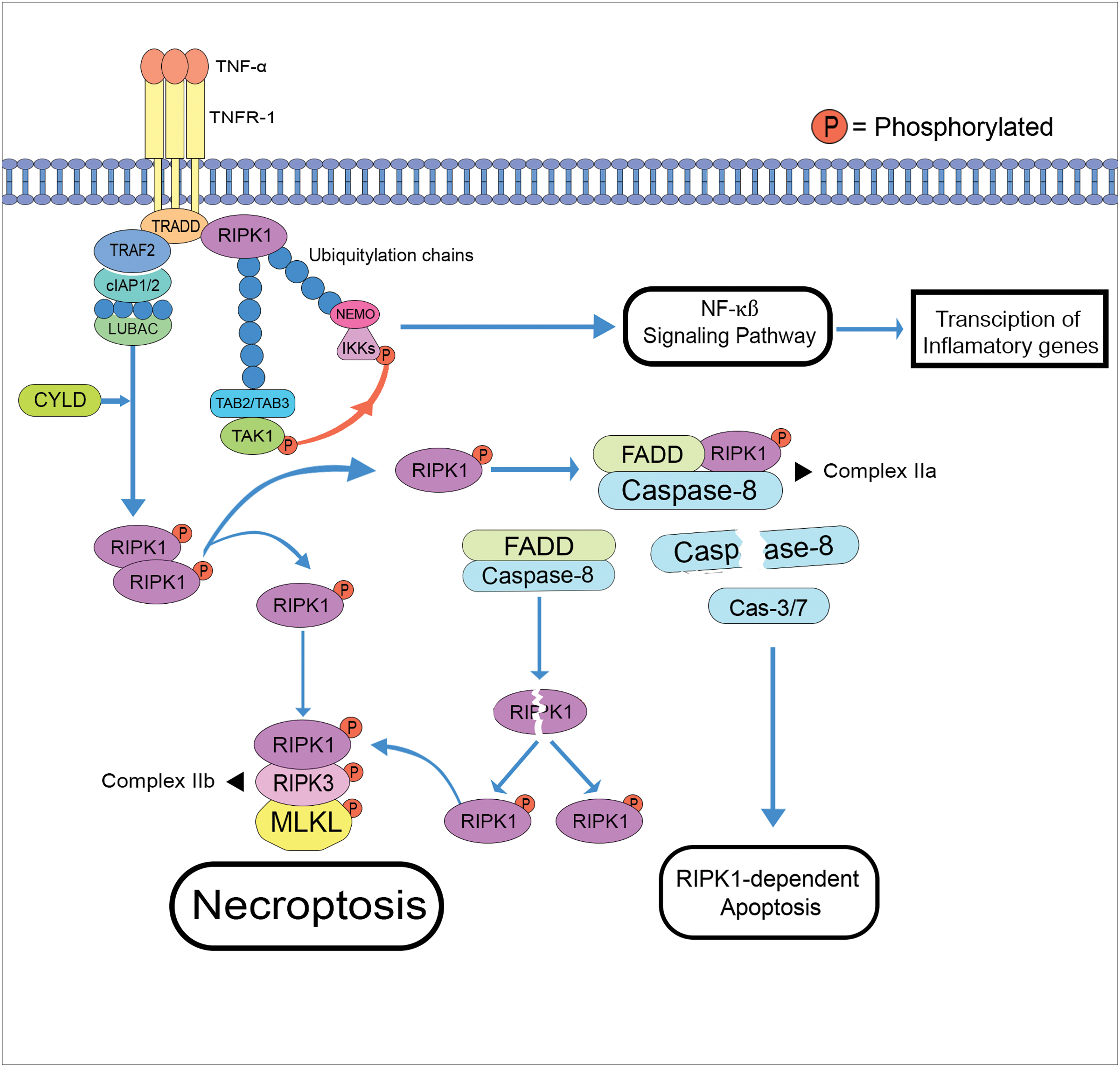
Figure 4: Schematic diagram of necroptosis and the RIPK1-dependent apoptosis pathways (cleavage of caspase-8 from complex IIa starts the caspase cascade) (TRADD: TNFR1-associated death domain; TRAF2: TNFR-associated factor-2; LUBAC: linear ubiquitination chain assembly complex; CYLD: cylindromatosis; TAB2/TAB3: TAK1-binding protein 2/TAK1-binding protein 3; TAK1: transforming growth factor-β-activated kinase; NEMO: NF-κB essential modulator; IKK: IκB kinase; FADD: fas-associated death domain; MLKL: mixed lineage kinase domain-Like protein).
RIPK1 in Acute Respiratory Distress Syndrome
Necroptosis in alveolar macrophages of virus-induced acute respiratory distress syndrome
Previous studies found that pro-inflammatory/cytotoxic alveolar macrophages are responsible for the damage and development of ARDS releasing TNF-α, IL-1β, IL-6, IL-12, IFNγ, etc., during infection for limiting the spread of viral loads (Laskin et al., 2019). For instance, the influenza A virus can elevate the accumulation and release of TNF-α mRNA, subsequently advancing to cell death, and the presence of free lipopolysaccharide (LPS) increases the production of this cytokine from the infected macrophages (Nain et al., 1990; Peschke et al., 1993; Bender et al., 1993). Further, the swine influenza virus (e.g., H1N1) induces the RIPK1 in porcine alveolar macrophage to phosphorylate the S579 location of dynamin-related protein 1 (DRP1), triggering the NLRP3 inflammasome activation. It also promotes IL-1β production inducing more inflammatory cytokine release and cell death via necroptosis and apoptosis (RIPK1–RIPK3–Caspase-8) (Kuriakose et al., 2016; Park et al., 2018). Faust and Mangalmurti (2020). have exhaustively reviewed that the formation of complex IIb (RIPK1-RIPK3-MLKL) and the RIPK1-independent TRIF-RIPK3, caused by bacterial and viral pulmonary infections generally directs the programmed cell death of the alveolar macrophages, along with the type I and type II alveolar epithelial cells. Further, the key molecules in complex IIb could either protect or prevent the replication of viral pneumonia, more specifically, the insulative role of RIPK3 to the transcriptional and post-transcriptional levels of some pathogens (Faust and Mangalmurti, 2020). Apart from the influenza virus, the respiratory syncytial virus (RSV) could also benignly induce ARDS (Hammer et al., 1997). Its binding receptors (RSV-G glycoprotein and RSV-F fusion glycoprotein) bind to the apical ciliated epithelial cells in the respiratory tract to induce intracellular replication activating the innate inflammatory immune response followed by necroptosis in the epithelial cells (Nam and Ison, 2019). This RSV-induced ARDS was supported by experiments conducted by research. For example, Santos et al. (2021) found activated RIPK1, RIPK3, and MLKL in both TNF-mediated mouse macrophage (Ripk3−/− mice) and human monocytes, which was promoted by RSV, confirming cell death of alveolar macrophage via necroptosis and in turn, damage the lung epithelial cells. In contrast, Simpson et al. (2020) reported that necroptosis was seen to promote viral load suppression by attenuating the RSV-induced HMGB1 translocation in pediatric donors of human aortic endothelial cells (hAEC). Hence, cases of necroptosis activity could be detrimental or beneficial depending on the conditions.
Role of RIPK1 in Virus-Induced Acute Respiratory Distress Syndrome
In general, RIPK1 is actively present during viral infections of ARDS since its elevated up-regulation along with RIPK3, caspase-3, caspase-8, and MLKL can be seen in almost all cases. This suggests that these listed inflammatory mediators are intimately involved in this syndrome (Pan et al., 2016). Furthermore, the extensive activation of RIPK1 molecule can be seen in deceased patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-induced COVID-19. For instance, Xu et al. (2021), from the Southern University of Science and Technology, Shenzhen, China, found high levels of pro-inflammatory cytokine transcripts in the bronchoalveolar lavage fluid (BALF), lung tissue, and peripheral blood mononuclear cells (PBMC). Additionally, a staggering number of activated RIPK1 in the lung epithelial cells was also mentioned in their study. Although the researchers found no evidence of necroptosis due to the lack of phosphorylated MLKL, apoptotic-mediated cell death of macrophage and epithelium was suggested due to the high amounts of detected cleaved-caspase-3 (c-casp3). Likewise, SARS-CoV-2 was identified to manipulate the RIPK-mediated inflammatory response of the host to its benefit in the angiotensin-converting enzyme-2 (ACE2+)-abundant type II pneumocytes (Xu et al., 2021). The RNA-dependent RNA polymerase (RdRp) (NSP12) protein is a key molecule for the replication of RNA-based viruses. Its RdRp domain (amino acid residues S367 to F920) and interface domain (amino acid residues A250 to R365) interact with the kinase domain of RIPK1 to activate it. This binding secures viral entry and survival through transcriptional induction of host factors such as the SARS-CoV-2 binding protein ACE2, epidermal growth factor receptor (EGFR), and certain inflammatory cytokines contributing more damage to the development of ARDS (Gordon et al., 2020; Xu et al., 2021; Pathania et al., 2022).
H7N9 is a sub-type of the influenza A virus (IAV) family that was identified around 2013 in China. Several infected patients had critical respiratory problems such as ARDS and organ failures (40% fatality rate), leading to their untimely death (Gao et al., 2013; Huang and Wang, 2020). Zhang et al. (2019) found that this avian IAV infects CD14+ monocyte on PBMCs and incessantly activates both the apoptosis and necroptosis pathway. Although the RIPK1 level was seen to be reduced 3 h post-infection, the 12-h mark showed its gradual increase, and necroptosis was still observed due to the rising level of RIPK3 and p-MLKL. Simultaneously, though treatment with Nec-1 and RIPK3 inhibitor (GSK’872 + GSK’843; GSKs) did not increase the cell viability levels of H7N9 infected monocytes, these inhibitors tremendously expunged the MLKL phosphorylation and possibly abrogated the necrotic pathway. Interestingly, another study found higher protein levels of RIPK1, RIPK3, and p-RIPK3 along with the MLKL and p-MLKL in H7N9-induced ARDS from deceased patients affirming cell death through necroptosis. They suspected that the formation of the necrosomal complex was due to the lack of cIAP2 expression and of its inhibition of RIPK1 (Qin et al., 2019). Another type of IAV known as H1N1 or the swine flu caused a massive public health emergency back in 2009. The flu caused 575,000 deaths and over 201,200 respiratory-related deaths globally (Dawood et al., 2012; Chauhan and Gordon, 2020). Like the H7N9 influenza A virus, people infected with the swine flu progressively developed ALI-ARDS. The virus was observed to up-regulate RIPK1 expressions in alveolar macrophages to induce IL-1β production, contributing to lung inflammation and damage (Park et al., 2018).
RIPK1 in Other Pathological Factor (Bacterial and External)-Induced Acute Respiratory Distress Syndrome
External factors and unhealthy lifestyle habits can also contribute to the development of ARDS. For example, air pollution, smoking, alcohol, and illegal substance abuse are among the few causes of the syndrome (Lin et al., 2018; Moazed et al., 2018). Cigarette smoke has been known to damage lung epithelial cells and walls of the alveoli. According to Pouwels et al. (2016), smoking can also induce necroptosis, and activate/release DAMPs prompting the innate immune system to respond to the site of cigarette smoke-induced necroptosis. The involvement of necroptosis provides a clue in the pivotal role of RIPK1 during cigarette smoke exposure. The researchers used Nec-1 inhibitor to confirm necroptosis in DAMP release and as a treatment to reduce neutrophilic inflammation. Other than mediating the activation and release of DAMPs, RIPK1 can activate the PARylated proteins generated by activated poly (ADP-ribose) polymerase 1 (PARP1) in the parthanatos pathway, which is the common pathway of programmed cell death in cigarette smoke exposure. Also known as the PARP1/apoptosis-inducing factor (AIF) (Greenwald and Pierce, 2019) signaling pathway, this form of cell death reacts to DNA damage caused by the inhalation of smoke and elevates the transcription of pro-inflammatory genes (Jang et al., 2018). Although the studies are limited regarding its interaction with PARP1, previous studies have shown evidence that the catalytic domain of PARP1 can interact and bind with RIPK1 during oxidative stress stimulation, which activates the mediator and triggers parthanatos cell death pathway (Jang et al., 2018; Künzi and Holt, 2019).
Apart from the external factors that cause ARDS, LPS from gram-negative bacteria is generally used to induce ALI/ARDS in murine models to study and study the disease. Such ALI/ARDS-based stimulation works since the LPS-TLR4 (lipopolysaccharide-Toll-like receptor 4) signaling pathway could also induce necroptosis in alveolar macrophages, which promotes the formation of toll/interleukin-1 receptor domain-containing adaptor protein (TIRAP) and toll/interleukin 1 receptor-domain-containing adapter-inducing interferon-β (TRIF) joined with the RIPK1-RIPK3-MLKL complex. However in the events of tissue damage, the caveolae-mediated TLR4 internalization prevents this complex formation decreasing the alveolar macrophage necroptosis (Chen et al., 2010; Fan and Fan, 2018). In previous studies, LPS was also shown to amplify inflammation in COVID-19-induced ARDS since it increases the ROS production, and activates the p38-MAPK pathway through interaction with CD14+ receptors, including the TLR4. Further, it generally amplifies the activation of the NF-κB pathway in THP-1 monocytic cells, these effects could also have lasting changes in pulmonary functions (Peschke et al., 1993; Tsikis et al., 2022). A report suggests that the apoptotic involvement of RIPK1 on the inflamed lung of a gram-negative bacterium Pasteurella multocida-infected chicken leads not only to necroptosis but also edema and hyperemia (Li et al., 2020). This was due to the activation of RIPK1-dependent apoptosis and initiation of the NF-κB signaling pathway causing inflammation of the alveolar pneumocytes (Pasparakis and Vandenabeele, 2015; Nežić et al., 2022). Hence, RIPK1 manages to display its presence and cell death capabilities in every aspect of non-pulmonary factors of ARDS, whether it is bacteremic sepsis or chronic airway inflammation.
This review discusses the role of RIPK1 and related apoptotic and necroptotic signaling pathways in the pathogenesis of ARDS. Many recent findings suggest that RIPK1 and related cell death machinery molecules might be potential targets for developing therapeutic strategies in treating ARDS. The death domain of RIPK1 initiates the formation of subsequent complexes and their pathways toward the survival or death of the cell. It is clear that RIPK1 recruitment to the TRADD starts the ubiquitylation and ligase inhibition of RIPK1, which leads to the NF-κB pathway, while its dissociation from this complex I leads to either apoptosis or necroptosis. While RIPK1-dependent apoptosis relies on the caspase cascade, i.e., the cleavage of caspase 8 to other caspases (3/7), necroptosis relies heavily on the MLKL phosphorylation. The major aspects of these signaling pathways in inflammatory diseases are well known. However, the function of RIPK1 is not restricted to caspase and MLKL activation alone, rather, it also interacts with many molecules, such as in the cases of virus-induced ARDS.
In COVID-19-induced ARDS, RIPK1-dependent apoptosis was often observed in lung epithelial cells and alveolar macrophages, suggesting that RIPK1 is present at high levels during the cytokine storm of COVID-19-induced ARDS, while its interaction with the RNA-dependent RNA polymerase (RdRp) domain ensures viral survivability and entry (Aftab et al., 2020). Further, in IAV-induced ARDS, the predominant presence of p-MLKL and p-RIPK3 determined that necroptosis was the mode of cell death in most cases (Xu et al., 2021). Moreover, the influenza swine virus also utilizes RIPK1 to phosphorylate the dynamin-related protein 1 (Drp1) at S579 to produce IL-1β cytokine through NLRP3 inflammasome activation and completely enhance lung inflammation by inducing cytokine production (Valera-Alberni et al., 2021). Necroptosis could also be induced in the alveolar macrophage in the lung epithelial cells, and provide advantageous survival of the RSV-induced ARDS, using its binding receptors to manipulate the macrophage into activating the immune response and completely destroy the epithelial cells. In several studies where necrostatin was used as an inhibitor of RIPK1, the pRIPK1-pRIPK3-MLKL complex was reduced, which led to the mitigation of necroptotic cell death (Pouwels et al., 2016; Lee et al., 2019). Nec-1s, as one of the first inhibitors and markers for RIPK1 has laid a substantial contribution to clinical trials of inflammatory diseases in this context (Yuan et al., 2019). Therefore, the acknowledgment of the functions of RIPK1 as a cell death mediator has been the subject of interest in the field of immunology. Further, the outstanding discovery of its inhibitor molecules, such as necrostatin, and GSKs, has made it a focal point for a potential therapeutic target for inflammatory diseases, including ALI/ARDS. Although there is still a need for an in-depth analysis of its mechanism and effect, the potentials for its use in treatment from studies are highly promising.
Acknowledgement: None.
Funding Statement: This study was supported by the National Natural Science Foundation of China under Grant 31970897, Outstanding Youth Foundation of Jiangsu Province (BK20190069, China), the Fundamental Research Funds for the Central Universities No. 30919011102 (China), and Qing Lan Project of Jiangsu Province, China.
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Xunan Zhao, Dan Weng, Emmanuel Mago; literature acquisition: Xunan Zhao, Emmanuel Mago; figures: Emmanuel Mago; draft manuscript preparation: Xunan Zhao, Emmanuel Mago; manuscript revision: Xunan Zhao, Dan Weng, Emmanuel Mago. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
Abdulnour R-EE, Gunderson T, Barkas I, Timmons JY, Barnig C et al. (2018). Early intravascular events are associated with development of acute respiratory distress syndrome. A substudy of the LIPS-A clinical trial. American Journal of Respiratory and Critical Care Medicine 197: 1575–1585. https://doi.org/10.1164/rccm.201712-2530OC [Google Scholar] [PubMed] [CrossRef]
Aeffner F, Bolon B, Davis IC (2015). Mouse models of acute respiratory distress syndrome. Toxicologic Pathology 43: 1074–1092. https://doi.org/10.1177/0192623315598399 [Google Scholar] [PubMed] [CrossRef]
Aftab SO, Ghouri MZ, Masood MU, Haider Z, Khan Z, Ahmad A, Munawar N (2020). Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. Journal of Translational Medicine 18: 275. https://doi.org/10.1186/s12967-020-02439-0 [Google Scholar] [PubMed] [CrossRef]
Akgul C, Moulding DA, Edwards SW (2001). Molecular control of neutrophil apoptosis. FEBS Letters 487: 318–322. https://doi.org/10.1016/S0014-5793(00)02324-3 [Google Scholar] [PubMed] [CrossRef]
Annibaldi A, Wicky John S, Vanden Berghe T, Swatek KN, Ruan J et al. (2018). Ubiquitin-mediated regulation of RIPK1 kinase activity independent of IKK and MK2. Molecular Cell 69: 566–580.e5. https://doi.org/10.1016/j.molcel.2018.01.027 [Google Scholar] [PubMed] [CrossRef]
Bakopoulos A, Kapelouzou A, Tsilimigras DI, Katsimpoulas M, Schizas D et al. (2017). Expression of Toll-like receptors (TLRs) in the lungs of an experimental sepsis mouse model. PLoS One 12: e0188050. https://doi.org/10.1371/journal.pone.0188050 [Google Scholar] [PubMed] [CrossRef]
Bates JHT, Smith BJ (2018). Ventilator-induced lung injury and lung mechanics. Annals of Translational Medicine 6: 378. https://doi.org/10.21037/atm.2018.06.29 [Google Scholar] [PubMed] [CrossRef]
Bender A, Sprenger H, Gong JH, Henke A, Bolte G, Spengler HP, Nain M, Gemsa D (1993). The potentiating effect of LPS on tumor necrosis factor-α production by influenza a virus-infected macrophages. Immunobiology 187: 357–371. https://doi.org/10.1016/S0171-2985(11)80350-5 [Google Scholar] [PubMed] [CrossRef]
Borriello F, Iannone R, Marone G (2017). Histamine release from mast cells and basophils. Histamine and Histamine Receptors in Health and Disease, pp. 121–139. https://doi.org/10.1007/164_2017_18 [Google Scholar] [PubMed] [CrossRef]
Butt Y, Kurdowska A, Allen TC (2016). Acute lung injury: A clinical and molecular review. Archives of Pathology & Laboratory Medicine 140: 345–350. https://doi.org/10.5858/arpa.2015-0519-RA [Google Scholar] [PubMed] [CrossRef]
Cao L, Mu W (2021). Necrostatin-1 and necroptosis inhibition: Pathophysiology and therapeutic implications. Pharmacological Research 163: 105297. https://doi.org/10.1016/j.phrs.2020.105297 [Google Scholar] [PubMed] [CrossRef]
Chauhan RP, Gordon ML (2020). A systematic review analyzing the prevalence and circulation of influenza viruses in swine population worldwide. Pathogens 9: 355. https://doi.org/10.3390/pathogens9050355 [Google Scholar] [PubMed] [CrossRef]
Chen H, Bai C, Wang X (2010). The value of the lipopolysaccharide-induced acute lung injury model in respiratory medicine. Expert Review of Respiratory Medicine 4: 773–783. https://doi.org/10.1586/ers.10.71 [Google Scholar] [PubMed] [CrossRef]
Chen KW, Demarco B, Ramos S, Heilig R, Goris M et al. (2021a). RIPK1 activates distinct gasdermins in macrophages and neutrophils upon pathogen blockade of innate immune signaling. Proceedings of the National Academy of Sciences 118: e2101189118. https://doi.org/10.1073/pnas.2101189118 [Google Scholar] [PubMed] [CrossRef]
Chen L, Tao W, Ji W, Lu Y, Zhao X (2021b). Effects of pulmonary fibrosis and surface tension on alveolar sac mechanics in diffuse alveolar damage. Journal of Biomechanical Engineering 143: 081013. https://doi.org/10.1115/1.4050789 [Google Scholar] [PubMed] [CrossRef]
Coldren CD, Nick JA, Poch KR, Woolum MD, Fouty BW et al. (2006). Functional and genomic changes induced by alveolar transmigration in human neutrophils. American Journal of Physiology-Lung Cellular and Molecular Physiology 291: L1267–L1276. https://doi.org/10.1152/ajplung.00097.2006 [Google Scholar] [PubMed] [CrossRef]
Cuny GD, Degterev A (2021). RIPK protein kinase family: Atypical lives of typical kinases. Seminars in Cell & Developmental Biology 109: 96–105. https://doi.org/10.1016/j.semcdb.2020.06.014 [Google Scholar] [PubMed] [CrossRef]
D’Alessio FR (2018). Mouse models of acute lung injury and ARDS. Methods in Molecular Biology 1809: 341–350. [Google Scholar] [PubMed]
D’Arcy MS (2019). Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biology International 43: 582–592. https://doi.org/10.1002/cbin.11137 [Google Scholar] [PubMed] [CrossRef]
Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK et al. (2012). Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: A modelling study. The Lancet Infectious Diseases 12: 687–695. https://doi.org/10.1016/S1473-3099(12)70121-4 [Google Scholar] [PubMed] [CrossRef]
Degterev A, Zhou W, Maki JL, Yuan J (2014). Assays for necroptosis and activity of RIP kinases. Methods in Enzymology 545: 1–33. https://doi.org/10.1016/B978-0-12-801430-1.00001-9 [Google Scholar] [PubMed] [CrossRef]
Dolcet X, Llobet D, Pallares J, Matias-Guiu X (2005). NF-κB in development and progression of human cancer. Virchows Archiv 446: 475–482. https://doi.org/10.1007/s00428-005-1264-9 [Google Scholar] [PubMed] [CrossRef]
Fan EKY, Fan J (2018). Regulation of alveolar macrophage death in acute lung inflammation. Respiratory Research 19: 50. https://doi.org/10.1186/s12931-018-0756-5 [Google Scholar] [PubMed] [CrossRef]
Faust H, Mangalmurti NS (2020). Collateral damage: Necroptosis in the development of lung injury. American Journal of Physiology-Lung Cellular and Molecular Physiology 318: L215–L225. https://doi.org/10.1152/ajplung.00065.2019 [Google Scholar] [PubMed] [CrossRef]
Friedrichson B, Mutlak H, Zacharowski K, Piekarski F (2021). Insight into ECMO, mortality and ARDS: A nationwide analysis of 45,647 ECMO runs. Critical Care 25: 38. https://doi.org/10.1186/s13054-021-03463-2 [Google Scholar] [PubMed] [CrossRef]
Gao R, Cao B, Hu Y, Feng Z, Wang D et al. (2013). Human infection with a novel avian-origin influenza A (H7N9) virus. New England Journal of Medicine 368: 1888–1897. https://doi.org/10.1056/NEJMoa1304459 [Google Scholar] [PubMed] [CrossRef]
Geng J, Ito Y, Shi L, Amin P, Chu J et al. (2017). Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nature Communications 8: 359. https://doi.org/10.1038/s41467-017-00406-w [Google Scholar] [PubMed] [CrossRef]
Goh QY, Kong A (2020). Videolaryngoscope-assisted double-lumen endotracheal tube intubation in an awake patient with known difficult airway and bronchopleural fistula: A case report. A & A Practice 14: e01186. https://doi.org/10.1213/XAA.0000000000001186 [Google Scholar] [PubMed] [CrossRef]
Gokey JJ, Snowball J, Sridharan A, Sudha P, Kitzmiller JA, Xu Y, Whitsett JA (2021). YAP regulates alveolar epithelial cell differentiation and AGER via NFIB/KLF5/NKX2-1. iScience 24: 102967. https://doi.org/10.1016/j.isci.2021.102967 [Google Scholar] [PubMed] [CrossRef]
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K et al. (2020). A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583: 459–468. https://doi.org/10.1038/s41586-020-2286-9 [Google Scholar] [PubMed] [CrossRef]
Gorman EA, O’Kane CM, McAuley DF (2022). Acute respiratory distress syndrome in adults: Diagnosis, outcomes, long-term sequelae, and management. The Lancet 400: 1157–1170. https://doi.org/10.1016/S0140-6736(22)01439-8 [Google Scholar] [PubMed] [CrossRef]
Greenwald SH, Pierce EA (2019). Parthanatos as a cell death pathway underlying retinal disease. Retinal Degenerative Diseases 1185: 323–327. https://doi.org/10.1007/978-3-030-27378-1_53 [Google Scholar] [PubMed] [CrossRef]
Hammer J, Numa A, Newth CJL (1997). Acute respiratory distress syndrome caused by respiratory syncytial virus. Pediatric Pulmonology 23: 176–183. https://doi.org/10.1002/(ISSN)1099-0496 [Google Scholar] [CrossRef]
He YQ, Zhou CC, Yu LY, Wang L, Deng J, Tao YL, Zhang F, Chen WS (2021). Natural product derived phytochemicals in managing acute lung injury by multiple mechanisms. Pharmacological Research 163: 105224. https://doi.org/10.1016/j.phrs.2020.105224 [Google Scholar] [PubMed] [CrossRef]
Herrera VLM, Walkey AJ, Nguyen MQ, Gromisch CM, Mosaddhegi JZ et al. (2022). A targetable ‘rogue’ neutrophil-subset, [CD11b+DEspR+] immunotype, is associated with severity and mortality in acute respiratory distress syndrome (ARDS) and COVID-19-ARDS. Scientific Reports 12: 5583. https://doi.org/10.1038/s41598-022-09343-1 [Google Scholar] [PubMed] [CrossRef]
Herrero R, Sanchez G, Lorente JA (2018). New insights into the mechanisms of pulmonary edema in acute lung injury. Annals of Translational Medicine 6: 32. https://doi.org/10.21037/atm.2017.12.18 [Google Scholar] [PubMed] [CrossRef]
Huang SW, Wang SF (2020). The effects of genetic variation on H7N9 avian influenza virus pathogenicity. Viruses 12: 1220. https://doi.org/10.3390/v12111220 [Google Scholar] [PubMed] [CrossRef]
Huppert L, Matthay M, Ware L (2019). Pathogenesis of acute respiratory distress syndrome. Seminars in Respiratory and Critical Care Medicine 40: 031–039. https://doi.org/10.1055/s-0039-1683996 [Google Scholar] [PubMed] [CrossRef]
Jang KH, Jang T, Son E, Choi S, Kim E (2018). Kinase-independent role of nuclear RIPK1 in regulating parthanatos through physical interaction with PARP1 upon oxidative stress. Biochimica et Biophysica Acta (BBA)—Molecular Cell Research 1865: 132–141. https://doi.org/10.1016/j.bbamcr.2017.10.004 [Google Scholar] [PubMed] [CrossRef]
Kanaoka Y, Austen KF (2019). Roles of cysteinyl leukotrienes and their receptors in immune cell-related functions. Advances in Immunology 42: 65–84. https://doi.org/10.1016/bs.ai.2019.04.002 [Google Scholar] [PubMed] [CrossRef]
Karhausen J, Choi HW, Maddipati KR, Mathew JP, Ma Q, Boulaftali Y, Lee RH, Bergmeier W, Abraham SN (2020). Platelets trigger perivascular mast cell degranulation to cause inflammatory responses and tissue injury. Science Advances 6: eaay6314. https://doi.org/10.1126/sciadv.aay6314 [Google Scholar] [PubMed] [CrossRef]
Koehler P, Cornely OA, Böttiger BW, Dusse F, Eichenauer DA et al. (2020). COVID-19 associated pulmonary aspergillosis. Mycoses 63: 528–534. https://doi.org/10.1111/myc.13096 [Google Scholar] [PubMed] [CrossRef]
Kuriakose T, Man SM, Subbarao Malireddi RK, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, Kanneganti TD (2016). ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Science Immunology 1: aag2045. https://doi.org/10.1126/sciimmunol.aag2045 [Google Scholar] [PubMed] [CrossRef]
Künzi L, Holt GE (2019). Cigarette smoke activates the parthanatos pathway of cell death in human bronchial epithelial cells. Cell Death Discovery 5: 127. https://doi.org/10.1038/s41420-019-0205-3 [Google Scholar] [PubMed] [CrossRef]
Laskin DL, Malaviya R, Laskin JD (2019). Role of macrophages in acute lung injury and chronic fibrosis induced by pulmonary toxicants. Toxicological Sciences 168: 287–301. https://doi.org/10.1093/toxsci/kfy309 [Google Scholar] [PubMed] [CrossRef]
Lee ACY, Zhang AJX, Chu H, Li C, Zhu H, Mak WWN, Chen Y, Kok KH, To KKW, Yuen KY (2019). H7N9 influenza A virus activation of necroptosis in human monocytes links innate and adaptive immune responses. Cell Death & Disease 10: 442. https://doi.org/10.1038/s41419-019-1684-0 [Google Scholar] [PubMed] [CrossRef]
Li L, Jin S, Zhang Y (2015). Ischemic preconditioning potentiates the protective effect of mesenchymal stem cells on endotoxin-induced acute lung injury in mice through secretion of exosome. International Journal of Clinical and Experimental Medicine 8: 3825–3832. [Google Scholar] [PubMed]
Li W, Li D, Chen Y, Abudou H, Wang H, Cai J, Wang Y, Liu Z, Liu Y, Fan H (2022). Classic signaling pathways in alveolar injury and repair involved in sepsis-induced ALI/ARDS: New research progress and prospect. Disease Markers 2022: v.2022. [Google Scholar]
Li W, Tang Q, Dai N, Feng W, Xie C, Cheng G, Liu X, Zhang W, Hu X, Gu C (2020). Receptor-interacting serine/threonine kinase 1- and 3-dependent inflammation induced in lungs of chicken infected with Pasteurella multocida. Scientific Reports 10: 6340. https://doi.org/10.1038/s41598-020-62042-7 [Google Scholar] [PubMed] [CrossRef]
Liaqat A, Mason M, Foster BJ, Kulkarni S, Barlas A et al. (2022). Evidence-based mechanical ventilatory strategies in ARDS. Journal of Clinical Medicine 11: 319. https://doi.org/10.3390/jcm11020319 [Google Scholar] [PubMed] [CrossRef]
Lin H, Tao J, Kan H, Qian Z, Chen A et al. (2018). Ambient particulate matter air pollution associated with acute respiratory distress syndrome in Guangzhou, China. Journal of Exposure Science & Environmental Epidemiology 28: 392–399. https://doi.org/10.1038/s41370-018-0034-0 [Google Scholar] [PubMed] [CrossRef]
Liu X, Tang AL, Chen J, Gao N, Zhang G, Xiao C (2023). RIPK1 in the inflammatory response and sepsis: Recent advances, drug discovery and beyond. Frontiers in Immunology 14: 1114103. https://doi.org/10.3389/fimmu.2023.1114103 [Google Scholar] [PubMed] [CrossRef]
Liu C, Xiao K, Xie L (2022). Advances in the use of exosomes for the treatment of ALI/ARDS. Frontiers in Immunology 13: 971189. https://doi.org/10.3389/fimmu.2022.971189 [Google Scholar] [PubMed] [CrossRef]
Liu X, Xie X, Ren Y, Shao Z, Zhang N, Li L, Ding X, Zhang L (2021). The role of necroptosis in disease and treatment. MedComm 2: 730–755. https://doi.org/10.1002/mco2.108 [Google Scholar] [CrossRef]
Maamar A, Delamaire F, Reizine F, Lesouhaitier M, Painvin B et al. (2023). Impact of arterial CO2 retention in patients with moderate or severe ARDS. Respiratory Care 68: 582–591. https://doi.org/10.4187/respcare.10507 [Google Scholar] [PubMed] [CrossRef]
Mackman N, Antoniak S, Wolberg AS, Kasthuri R, Key NS (2020). Coagulation abnormalities and thrombosis in patients infected with SARS-CoV-2 and other pandemic viruses. Arteriosclerosis, Thrombosis, and Vascular Biology 40: 2033–2044. https://doi.org/10.1161/ATVBAHA.120.314514 [Google Scholar] [PubMed] [CrossRef]
Malireddi RKS, Kesavardhana S, Karki R, Kancharana B, Burton AR, Kanneganti TD (2020). RIPK1 distinctly regulates yersinia-induced inflammatory cell death, PANoptosis. ImmunoHorizons 4: 789–796. https://doi.org/10.4049/immunohorizons.2000097 [Google Scholar] [PubMed] [CrossRef]
Matthay MA, Ware LB, Zimmerman GA (2012). The acute respiratory distress syndrome. Journal of Clinical Investigation 122: 2731–2740. https://doi.org/10.1172/JCI60331 [Google Scholar] [PubMed] [CrossRef]
Meng H, Liu Z, Li X, Wang H, Jin T et al. (2018). Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proceedings of the National Academy of Sciences 115: E2001–E2009. https://doi.org/10.1073/pnas.1722013115 [Google Scholar] [PubMed] [CrossRef]
Meyer NJ, Gattinoni L, Calfee CS (2021). Acute respiratory distress syndrome. The Lancet 398: 622–637. https://doi.org/10.1016/S0140-6736(21)00439-6 [Google Scholar] [PubMed] [CrossRef]
Moazed F, Hendrickson C, Nelson M, Conroy A, Cohen MJ, Calfee CS (2018). Platelet aggregation after blunt trauma is associated with the acute respiratory distress syndrome and altered by cigarette smoke exposure. Journal of Trauma and Acute Care Surgery 84: 365–371. https://doi.org/10.1097/TA.0000000000001738 [Google Scholar] [PubMed] [CrossRef]
Mokra D (2020). Acute lung injury—From pathophysiology to treatment. Physiological Research 69: S353–S366. [Google Scholar] [PubMed]
Nain M, Hinder F, Gong JH, Schmidt A, Bender A, Sprenger H, Gemsa D (1990). Tumor necrosis factor-alpha production of influenza A virus-infected macrophages and potentiating effect of lipopolysaccharides. Journal of Immunology 145: 1921–1928. https://doi.org/10.4049/jimmunol.145.6.1921 [Google Scholar] [CrossRef]
Nam HH, Ison MG (2019). Respiratory syncytial virus infection in adults. BMJ 66: l5021. https://doi.org/10.1136/bmj.l5021 [Google Scholar] [PubMed] [CrossRef]
Nežić L, Amidžić L, Škrbić R, Gajanin R, Mandić D, Dumanović J, Milovanović Z, Jaćević V (2022). Amelioration of endotoxin-induced acute lung injury and alveolar epithelial cells apoptosis by simvastatin is associated with up-regulation of Survivin/NF-κB/p65 pathway. International Journal of Molecular Sciences 23: 2596. https://doi.org/10.3390/ijms23052596 [Google Scholar] [PubMed] [CrossRef]
Ofengeim D, Yuan J (2013). Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nature Reviews Molecular Cell Biology 14: 727–736. https://doi.org/10.1038/nrm3683 [Google Scholar] [PubMed] [CrossRef]
Ohshimo S (2021). Oxygen administration for patients with ARDS. Journal of Intensive Care 9: 17. https://doi.org/10.1186/s40560-021-00532-0 [Google Scholar] [PubMed] [CrossRef]
Olajuyin AM, Zhang X, Ji HL (2019). Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discovery 5: 63. https://doi.org/10.1038/s41420-019-0147-9 [Google Scholar] [PubMed] [CrossRef]
Pallister I (2005). Current concepts of the inflammatory response after major trauma: An update. Injury 36: 227–229. https://doi.org/10.1016/j.injury.2004.01.007 [Google Scholar] [PubMed] [CrossRef]
Pan L, Yao DC, Yu YZ, Chen BJ, Li SJ et al. (2016). Activation of necroptosis in a rat model of acute respiratory distress syndrome induced by oleic acid. Sheng Li Xue Bao (Acta physiologica Sinica) 68: 661–668. [Google Scholar] [PubMed]
Papadopoulos MC, Saadoun S, Verkman AS (2008). Aquaporins and cell migration. Pflügers Archiv—European Journal of Physiology 456: 693–700. https://doi.org/10.1007/s00424-007-0357-5 [Google Scholar] [PubMed] [CrossRef]
Papazian L, Aubron C, Brochard L, Chiche JD, Combes A et al. (2019). Formal guidelines: Management of acute respiratory distress syndrome. Annals of Intensive Care 9: 69. https://doi.org/10.1186/s13613-019-0540-9 [Google Scholar] [PubMed] [CrossRef]
Paris AJ, Hayer KE, Oved JH, Avgousti DC, Toulmin SA et al. (2020). STAT3-BDNF–TrkB signalling promotes alveolar epithelial regeneration after lung injury. Nature Cell Biology 22: 1197–1210. https://doi.org/10.1038/s41556-020-0569-x [Google Scholar] [PubMed] [CrossRef]
Park HS, Liu G, Liu Q, Zhou Y (2018). Swine influenza virus induces RIPK1/DRP1-mediated interleukin-1 beta production. Viruses 10: 419. https://doi.org/10.3390/v10080419 [Google Scholar] [PubMed] [CrossRef]
Pasparakis M, Vandenabeele P (2015). Necroptosis and its role in inflammation. Nature 517: 311–320. https://doi.org/10.1038/nature14191 [Google Scholar] [PubMed] [CrossRef]
Pathania S, Rawal RK, Singh PK (2022). RdRp (RNA-dependent RNA polymeraseA key target providing anti-virals for the management of various viral diseases. Journal of Molecular Structure 1250: 131756. https://doi.org/10.1016/j.molstruc.2021.131756 [Google Scholar] [PubMed] [CrossRef]
Peschel G, Jung EM, Fisser C, Putz FJ, Wertheimer T, Sinner B, Lunz D, Jung F, Müller M (2021). Interstitial lung opacities in patients with severe COVID-19 pneumonia by bedside high-resolution ultrasound in association to CO2 retention. Clinical Hemorheology and Microcirculation 77: 355–365. https://doi.org/10.3233/CH-200925 [Google Scholar] [PubMed] [CrossRef]
Peschke T, Bender A, Nain M, Gemsa D (1993). Role of macrophage cytokines in influenza A virus lnfections. Immunobiology 189: 340–355. https://doi.org/10.1016/S0171-2985(11)80365-7 [Google Scholar] [PubMed] [CrossRef]
Pooladanda V, Thatikonda S, Bale S, Pattnaik B, Sigalapalli DK, Bathini NB, Singh SB, Godugu C (2019). Nimbolide protects against endotoxin-induced acute respiratory distress syndrome by inhibiting TNF-α mediated NF-κB and HDAC-3 nuclear translocation. Cell Death & Disease 10: 81. https://doi.org/10.1038/s41419-018-1247-9 [Google Scholar] [PubMed] [CrossRef]
Potey PM, Rossi AG, Lucas CD, Dorward DA (2019). Neutrophils in the initiation and resolution of acute pulmonary inflammation: Understanding biological function and therapeutic potential. The Journal of Pathology 247: 672–685. https://doi.org/10.1002/path.5221 [Google Scholar] [PubMed] [CrossRef]
Pouwels SD, Zijlstra GJ, van der Toorn M, Hesse L, Gras R et al. (2016). Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. American Journal of Physiology-Lung Cellular and Molecular Physiology 310: L377–L386. https://doi.org/10.1152/ajplung.00174.2015 [Google Scholar] [PubMed] [CrossRef]
Qadri M, Jay GD, Zhang LX, Wong W, Reginato AM, Sun C, Schmidt TA, Elsaid KA (2018). Recombinant human proteoglycan-4 reduces phagocytosis of urate crystals and downstream nuclear factor kappa B and inflammasome activation and production of cytokines and chemokines in human and murine macrophages. Arthritis Research & Therapy 20: 192. https://doi.org/10.1186/s13075-018-1693-x [Google Scholar] [PubMed] [CrossRef]
Qin C, Sai X, Qian X, Wu Y, Zou L, Wang H, Bian T, Yan Z (2019). Close relationship between cIAP2 and human ARDS induced by severe H7N9 infection. BioMed Research International 2019: 1–9. https://doi.org/10.1155/2019/2121357 [Google Scholar] [PubMed] [CrossRef]
Rahmel T, Nowak H, Rump K, Siffert W, Peters J, Adamzik M (2018). The aquaporin 5-1364A/C promoter polymorphism impacts on resolution of acute kidney injury in pneumonia evoked ARDS. PLoS One 13: e0208582. [Google Scholar] [PubMed]
Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS (2012). Acute respiratory distress syndrome: The Berlin definition. Journal of the American Medical Association 307: 2526–2533. [Google Scholar] [PubMed]
Roh JS, Sohn DH (2018). Damage-associated molecular patterns in inflammatory diseases. Immune Network 18: e27. https://doi.org/10.4110/in.2018.18.e27 [Google Scholar] [PubMed] [CrossRef]
Safa RA (2013). Roles of c-FLIP in apoptosis, necroptosis, and autophagy. Journal of Carcinogenesis & Mutagenesis: 003. https://doi.org/10.4172/2157-2518.S6-003 [Google Scholar] [PubMed] [CrossRef]
Saguil A, Fargo MV (2020). Acute respiratory distress syndrome: Diagnosis and management. American Family Physician 101: 730–738. [Google Scholar] [PubMed]
Sahetya SK, Goligher EC, Brower RG (2017). Fifty years of research in ARDS. Setting positive end-expiratory pressure in acute respiratory distress syndrome. American Journal of Respiratory and Critical Care Medicine 195: 1429–1438. https://doi.org/10.1164/rccm.201610-2035CI [Google Scholar] [PubMed] [CrossRef]
Santos LD, Antunes KH, Muraro SP, de Souza GF, da Silva AG et al. (2021). TNF-mediated alveolar macrophage necroptosis drives disease pathogenesis during respiratory syncytial virus infection. European Respiratory Journal 57: 2003764. https://doi.org/10.1183/13993003.03764-2020 [Google Scholar] [PubMed] [CrossRef]
Sauler M, Bazan IS, Lee PJ (2019). Cell death in the lung: The apoptosis-necroptosis axis. Annual Review of Physiology 81: 375–402. [Google Scholar] [PubMed]
Simpson J, Loh Z, Ullah MA, Lynch JP, Werder RB et al. (2020). Respiratory syncytial virus infection promotes necroptosis and HMGB1 release by airway epithelial cells. American Journal of Respiratory and Critical Care Medicine 201: 1358–1371. https://doi.org/10.1164/rccm.201906-1149OC [Google Scholar] [PubMed] [CrossRef]
Song J, Yao L, Zhao L, Du B, Liu L, Chen J (2020). Changes in the concentrations of mediators in exhaled breath condensate during cardiac valve replacement under cardiopulmonary bypass and their relations with postoperative acute respiratory distress syndrome. Medicine 99: e20007. https://doi.org/10.1097/MD.0000000000020007 [Google Scholar] [PubMed] [CrossRef]
Tan Y, Chen Q, Li X, Zeng Z, Xiong W, Li G, Li X, Yang J, Xiang B, Yi M (2021). Pyroptosis: A new paradigm of cell death for fighting against cancer. Journal of Experimental & Clinical Cancer Research 40: 153. https://doi.org/10.1186/s13046-021-01959-x [Google Scholar] [PubMed] [CrossRef]
Tsikis ST, Fligor SC, Hirsch TI, Pan A, Yu LJ, Kishikawa H, Joiner MM, Mitchell PD, Puder M (2022). Lipopolysaccharide-induced murine lung injury results in long-term pulmonary changes and downregulation of angiogenic pathways. Scientific Reports 12: 10245. https://doi.org/10.1038/s41598-022-14618-8 [Google Scholar] [PubMed] [CrossRef]
Valera-Alberni M, Joffraud M, Miro-Blanch J, Capellades J, Junza A et al. (2021). Crosstalk between Drp1 phosphorylation sites during mitochondrial remodeling and their impact on metabolic adaptation. Cell Reports 36: 109565. https://doi.org/10.1016/j.celrep.2021.109565 [Google Scholar] [PubMed] [CrossRef]
Vassiliou AG, Manitsopoulos N, Kardara M, Maniatis NA, Orfanos SE, Kotanidou A (2017). Differential expression of aquaporins in experimental models of acute lung injury. In Vivo 31: 885–894. [Google Scholar] [PubMed]
Walkey A, Summer H, Ho V, Alkana P (2012). Acute respiratory distress syndrome: Epidemiology and management approaches. Clinical Epidemiology 4: 159–169. https://doi.org/10.2147/CLEP.S28800 [Google Scholar] [PubMed] [CrossRef]
Williams AE, José RJ, Mercer PF, Brealey D, Parekh D, Thickett DR, O’Kane C, McAuley DF, Chambers RC (2017). Evidence for chemokine synergy during neutrophil migration in ARDS. Thorax 72: 66–73. https://doi.org/10.1136/thoraxjnl-2016-208597 [Google Scholar] [PubMed] [CrossRef]
Xie YP, Chen CP, Wang JC, Qian GS, Wang YD, Xiao ZL (2005). Experimental study on the expression and function of aquaporin-1 and aquaporin-5 in rats with acute lung injury induced by lipopolysaccharide. Zhonghua Jie He He Hu Xi Za Zhi = Chinese Journal of Tuberculosis and Respiratory Diseases 28: 385–389 (In Chinese). [Google Scholar] [PubMed]
Xu G, Li Y, Zhang S, Peng H, Wang Y et al. (2021). SARS-CoV-2 promotes RIPK1 activation to facilitate viral propagation. Cell Research 31: 1230–1243. https://doi.org/10.1038/s41422-021-00578-7 [Google Scholar] [PubMed] [CrossRef]
Yadav E, Yadav N, Hus A, Yadav JS (2020). Aquaporins in lung health and disease: Emerging roles, regulation, and clinical implications. Respiratory Medicine 174: 106193. [Google Scholar] [PubMed]
Yang CY, Chen CS, Yiang GT, Cheng YL, Yong SB, Wu MY, Li CJ (2018). New insights into the immune molecular regulation of the pathogenesis of acute respiratory distress syndrome. International Journal of Molecular Sciences 19: 588. [Google Scholar] [PubMed]
Yin K, Wang S, Zhao RC (2019). Exosomes from mesenchymal stem/stromal cells: A new therapeutic paradigm. Biomarker Research 7: 8. [Google Scholar] [PubMed]
Yuan J, Amin P, Ofengeim D (2019). Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nature Reviews Neuroscience 20: 19–33. [Google Scholar] [PubMed]
Zeng Y, Xu D, Wei J, Yu H, Yang J, Yu S, Sun H (2021). Lidocaine pretreatment up-regulates aquaporin-5 expression in primary alveolar epithelium type II cells injured by lipopolysaccharides. Pakistan Journal of Pharmaceutical Sciences 34: 1699–1705. [Google Scholar] [PubMed]
Zhang X, Zhang H, Xu C, Li X, Li M et al. (2019). Ubiquitination of RIPK1 suppresses programmed cell death by regulating RIPK1 kinase activation during embryogenesis. Nature Communications 10: 4158. https://doi.org/10.1038/s41467-019-11839-w [Google Scholar] [PubMed] [CrossRef]
Zheng Y, Huang Y, Xu Y, Sang L, Liu X, Li Y (2023). Ferroptosis, pyroptosis and necroptosis in acute respiratory distress syndrome. Cell Death Discovery 9: 91. https://doi.org/10.1038/s41420-023-01369-2 [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools